1: Departamento de Ingeniería Geológica y Minera, Escuela Universitaria Politécnica de Almadén, Universidad de Castilla-La Mancha, Plaza M. Meca 1, 13400 Almadén, España.
2: Departamento de Cristalografía y Mineralogía, Facultad de Ciencias Geológicas, Universidad Complutense, 28040 Madrid, España.

Problemas potenciales derivados de la minería. En primer plano, estanque de soluciones cianuradas, atrás, pila de lixiviación. Punitaqui (Chile), explotación minera abandonada de Cu-Hg-Au
El tema ambiental está ganado importancia de manera progresiva en las Ciencias de la Tierra, tanto en la enseñanza como en la investigación teórica y aplicada. Aunque los aspectos más paisajísticos de los problemas ambientales suelen a veces llamar más la atención de la opinión pública (impactos visuales), existen otros, de fondo, que imprescindiblemente “deben” ser tratados. Nos referimos a la migración de metales y compuestos químicos en el ciclo exógeno. Estos procesos son interactivos, y toman lugar en la atmósfera, la hidrósfera, y en esa delgada y vulnerable “piel” que cubre gran parte de los continentes: los suelos. Los principales peligros ambientales a que se enfrenta la sociedad vienen dados por la extrema toxicidad, a determinadas concentraciones, de los llamados metales pesados y compuestos químicos, ya sean de origen natural o antropogénico. Incidentes de contaminación por mercurio, como el de la bahía de Minamata en Japón, que dejó todo un legado de nacimientos de niños deformes (teratogénesis) son un ejemplo de la importancia de este tema.
En este curso trataremos dos aspectos básicos de la geología ambiental, referidos normalmente de manera algo difusa como mineralogía ambiental y geoquímica ambiental. Generalmente la opinión pública suele fijar sus críticas sobre los impactos ambientales de origen industrial más evidentes, esto es (por ejemplo), de la presencia en si de una mina (impacto visual), olvidando los aspectos mineralógicos y químicos que se derivan de la actividad minera como tal. Aquí hay varios temas que comentar y analizar: la “mineralogía” del yacimiento que se explota, el o los “metales” presentes en esas fases minerales, los “procesos metalúrgicos” que se emplean para extraer el metal o metales, y los efectos del “clima” de una región sobre las variables anteriores. Obviamente no es lo mismo bajo el punto de vista de la salud humana y ambiental una explotación minera de hierro, que otra de arsénico o plomo. La primera podrá generar importantes impactos visuales o sociales, pero el hierro definitivamente no está dentro de la lista de elementos químicos de alta peligrosidad. De esta manera, el problema debe ser enfocado primariamente en términos de la mineralogía del yacimiento que ha sido explotado o se encuentra en explotación, continuando con los aspectos químicos derivados, analizando finalmente el problema bajo una perspectiva ambiental más amplia.
Así, estudiaremos a lo largo de este curso los dos aspectos más críticos la geología ambiental. Uno referido a los minerales, en sus dos vertientes: 1) como agentes de contaminación, y 2) en su aspecto “amable” esto es, en la resolución de problemas ambientales. Muchos minerales son solubles, esto es, bajo determinadas condiciones físico-químicas liberan su carga metálica. La introducción de estos metales o sales al ciclo exógeno la examinaremos bajo la óptica de la geoquímica. Al respecto cabe destacar que hay todo un camino de ida y vuelta entre estas dos visiones del problema. No podemos plantear un estudio geoquímico sin la adecuada comprensión del tema mineralógico, y por su parte, la mera mineralogía (sin apoyo de la geoquímica) nos dirá poco sobre el problema ambiental.
El curso que ofrecemos a continuación está estructurado de la siguiente manera:
1) Química de las soluciones en la naturaleza: definiendo conceptos básicos. Una guía breve y práctica sobre la química básica que necesitará para este curso.
2) Solubilización, transporte, y precipitación de substancias en el ciclo exógeno: una aproximación al tema de los contaminantes. Los elementos pasan de unos medios a otros en relación con procesos que están en función de la físico-química del medio y de sus propias características. Los procesos medioambientales se pueden caracterizar como procesos geológicos externos que ocurren de forma especialmente rápida. La geoquímica de estos procesos, en distintos medios: ambiente minero, industrial, las interacciones entre agua y minerales en ríos y lagos, o en relación con la atmósfera urbana, tienen un gran interés para identificar y valorar adecuadamente los procesos medioambientales.
3) Mineralogía y procesos de contaminación de suelos. El suelo es el receptor de la mayor parte de los residuos generados por el hombre, lo que produce su contaminación. La mineralogía del suelo es un parámetro fundamental para comprender las interacciones que aquí pueden producirse, y analizar la posibilidad de que la contaminación se transmita a otros recursos, como las aguas subterráneas, o a las cadenas tróficas, a través de su captación por las plantas.
4) Prospección geoquímica. La prospección geoquímica es una herramienta fundamental para identificar y valorar una contaminación ambiental, ya sea para reconocer el alcance y extensión de un problema conocido (por ejemplo, la contaminación inducida por una mina en su entorno) como para buscar las fuentes de una contaminación de origen desconocido.
5) Mineralogía y residuos mineros. Analizaremos los procesos mineralógicos que resultan de interés medioambiental en este campo.
6) Mineralogía y almacenamiento de residuos. El almacenamiento de residuos de todo tipo supone en la mayor parte de los casos su enterramiento controlado, y en este sentido, la naturaleza de los materiales que se emplean en este proceso tiene una gran importancia para asegurar un sellado de los almacenes.
7) Mineralogía y “mal de la piedra”. La acción de la intemperie sobre las edificaciones o sus complementos supone una degradación de las mismas, que será más rápida o más lenta en función de tres factores: el clima, la composición de la atmósfera, y la naturaleza de las rocas afectadas, y de su mineralogía en particular.
8) Minerales, metales, gases y la salud humana y ambiental. Los minerales a menudo entran en nuestro organismo, en unos casos por ingestión junto con los alimentos, en otros casos a través de la respiración, y pueden producir efectos por lo general nocivos para la salud. Así por ejemplo, el polvo de sílice ocasiona la enfermedad denominada silicosis, y los asbestos, la asbestosis. Por otra parte, los minerales pueden alterarse químicamente liberando su contenido metálico, el que puede tener en ocasiones efectos muy tóxicos para la salud humana y ambiental.
9) Minerales de interés en el control de procesos ambientales. Algunas minerales se están empleando o pueden tener aplicaciones futuras en el control de procesos ambientales. Así por ejemplo las ceolitas se usan para purificar agua, tanto en procesos industriales como domésticos.
Además, ofrecemos dos casos prácticos:
2.- Mercurio en el medio ambiente del Distrito de Almadén. Un fenómeno dinámico.
1.- Química de las soluciones en la naturaleza…
1.- Química de las soluciones en la naturaleza: definiendo conceptos básicos
Los procesos de contaminación en la naturaleza están íntimamente ligados a la hidrólisis, oxidación, o reducción de compuestos, el transporte de cationes y aniones o de substancias coloidales, y a la formación de nuevos compuestos químicos. En este sentido es importante entender las bases que rigen dichos procesos para poder predecir el comportamiento último de los posibles metales o substancias contaminantes.
Esquema del Tema:
1.1.- Expresando la concentración en las soluciones
1.2.- Definiendo ácido y base
1.3.- Equilibrio químico
1.4.- Hidrólisis
1.5.- Oxidación – reducción, diagramas Eh-pH
1.6.- Bibliografía
1.1.- Expresando la concentración en las soluciones
En soluciones denominamos soluto a la substancia que se disolverá en el solvente (comúnmente: agua). Existen múltiples maneras de expresar el grado de disolución del soluto, y aquí solo revisaremos dos de las más comunes: 1) la molaridad (M); y 2) la molalidad (m).
La molaridad se expresa en moles de soluto contenidos en 1 litro de solución. Por ejemplo, una solución 0.5 molar (0.5 M) de H2SO4 contiene 49.04 g de H2SO4. Esto es así ya que el peso molecular del ácido sulfúrico es de 98.08. En otras palabras:
![]()
La molalidad se expresa en moles de soluto por kilos de solvente. Por ejemplo, una solución constituida por 98.08 g de H2SO4 (1 mol) y 1000 g de agua tendrá una molalidad de 1 m (1 molal). En otras palabras:
![]()
1.2.- Definiendo ácido y base
Arrhenius propuso a finales del siglo XIX que los ácidos difieren de otras substancias que contienen hidrógeno en que éstos se disocian parcialmente en el agua, dando hidrogeniones libres (H+), por ejemplo:
HCl → H+ + Cl–
Sin embargo, Brønsted en el siglo XX, indicó que esta idea no podía ser correcta, ya que H+ no representa nada más que un protón aislado, el cual no puede existir como tal en presencia de agua. Más bien ocurriría algo como:
HCl + H2O → H3O+ + Cl–
Brønsted definió ácido como: una molécula o ión capaz de dar H+ a otra molécula o ión. En otras palabras un ácido sería un donante de protones. Otros químicos objetaron posteriormente también la idea de Brønsted por restrictiva. En lo que se refiere a este curso, utilizaremos la definición de Krauskopf (próxima a la de Arrhenius), que dice: ácido es una substancia que contiene hidrógeno y entrega iones de hidrógenos libre (H+) cuando se disuelve en agua.
El término base también ha tenido una historia larga. Por mucho tiempo se utilizó de manera ambigua para describir aquellas substancias que al ser disueltas presentaban: 1) un tacto jabonoso; 2) un sabor amargo; 3) la capacidad de neutralizar un ácido; y 4) la capacidad de revertir los cambios de colores que producían los ácidos en los tintes vegetales. Desde los tiempos de Arrhenius las propiedades de las bases han sido adscritas al ión oxidrilo (OH–), y el término se restringió a compuestos tales como Na(OH) o Ca(OH)2, los que se disocian y dan este ión directamente:
Ca(OH)2 → Ca2+ + 2 OH–
Brønsted se refiere al ión OH– como una base en si mismo, y amplía el término para incluir todas los iones o moléculas, que como el OH–, son capaces de unirse con H+ (substancias receptoras de protones). A efectos de este curso utilizaremos de nuevo la definición de Krauskopf, que dice: base es una substancia que contiene el grupo OH y entrega iones OH– cuando se disuelve en agua.
1.3.- Equilibrio Químico
En teoría todas las reacciones químicas son reversibles, es decir existe la posibilidad de que los compuestos resultantes reaccionen entre ellos para dar lugar a los reactantes originales:
A + B → C + D
C + D → A + B
Todo esto depende de la fuerza determinante (variación del potencial químico) que acompaña a una reacción, y constituye una medida exacta de la tendencia a que una reacción se complete. Así, si el valor absoluto de ésta es muy grande, y el signo negativo, la reacción se completará en el sentido directo de la misma:
A + B → C + D
Sin embargo, si el valor es solo ligeramente negativo, la reacción puede progresar hasta cierto punto, pero podría revertirse si las condiciones fisicoquímicas del sistema son ligeramente modificadas. En este caso diremos que la reacción es reversible:
A + B ↔ C + D
Si se cumplen las siguientes condiciones: 1) la velocidad de formación de los reactantes (A, B) es igual que la de los productos (C, D); 2) estos últimos reaccionan entre si para dar lugar a los reactantes; y 3) que la composición de la solución es constante; debemos decir entonces que la reacción es dinámica y constantemente reactiva en las dos direcciones, y por lo tanto que existe equilibrio químico:
A + B = C + D
La característica más importante del equilibrio químico es que la velocidad de reacción en las dos direcciones es igual, equilibrándose mutuamente, evitando así cualquier variación composicional en el sistema. La constante de equilibrio para una reacción es proporcional a las concentraciones de las substancias presentes en el sistema, y se expresa como:
![]()
donde los paréntesis cuadrados expresan la concentración química de las substancias (por ejemplo, en moles por litro) que intervienen en el equilibrio. Si en la reacción intervienen más de dos substancias:
A + B + C = D + E
la constante de equilibrio queda definida como:
![]()
Por otra parte, si intervienen dos o más moléculas de una substancia:
A + 3 B = 2 C
tendremos:
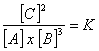
Y en el caso general:
aA + bB = cC + dD
tendremos:
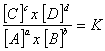
Para entender como funciona la constante de equilibrio nos centraremos ahora en dos temas importantes: 1) el producto de solubilidad, y 2) la disociación de ácidos y bases. El producto de solubilidad (Ksp, medido para las reacciones a 25ºC) nos da una medida de “cuanto” de un compuesto soluble o ligeramente soluble pasa efectivamente a fase iónica. Analizaremos dos ejemplos concretos, la fluorita (CaF2) y la anhidrita (CaSO4). La fluorita es un mineral típico de ganga en muchas masas filonianas, y en algunos casos es explotada como tal. Se trata de un compuesto iónico muy poco soluble en agua. La reacción de disolución queda expresada como:
CaF2 = Ca2+ + 2 F–
La constante de equilibrio para la reacción será:
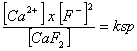 , de aquí en adelante expresado como KCaF2
, de aquí en adelante expresado como KCaF2
Debido a que CaF2 es una fase sólida tenemos que:
KCaF2 = [Ca2+] x [F–]2
0.017 g de CaF2 se disolverán completamente en 1 kilo de agua a 25ºC. Dado que la masa molecular de la fluorita es de 78.08, diremos que la molalidad es de 0.00022 mol/kg (0.017/78.08 = 0.00022). Si 0.00022 moles de CaF2 pueden saturar 1 kg de agua a 25ºC las concentraciones de Ca2+ y F– serán de 0.00022 y 0.00044 respectivamente, y así:
KCaF2 = [Ca2+] x [F–]2
= 0.00022 x (0.00044)2
= 4.26 x 10-11
Este producto de solubilidad muy bajo, y de ahí podemos extraer dos conclusiones: 1) que la fluorita se disuelve muy poco en solución acuosa; y 2) que en una solución basta con un ligero incremento en la concentración de iones Ca2+ o F– para que empiece a precipitar CaF2. Dado que los valores del Ksp vienen dados en tablas (no hace falta realizar los cálculos anteriores), podemos así predecir de una manera relativamente fácil el comportamiento en solución de un determinado compuesto iónico.
En el caso de la anhidrita la disolución procede de la siguiente manera:
CaSO4 = Ca2+ + SO4-2
y el producto de solubilidad vienen expresado como:
![]()
KCaSO4 = [Ca2+] x [SO4-2]
y el valor de este Ksp es de 10-4.5. Si comparamos los Ksp de la fluorita y la anhidrita (4.26 x 10-11 versus 10-4.5), veremos que esta última es mucho más soluble en solución acuosa que la primera, o que si comenzamos a subir la concentración de Ca2+ en una solución no saturada que contenga aniones F– y SO4-2, el primer mineral en empezar a precipitar será la fluorita.
El equilibrio químico también es importante para entender el grado de disociación que pueden presentar diferentes ácidos. Hablamos en este caso de la constante de disociación del ácido o Ka. Como ya hemos visto anteriormente, los ácidos inorgánicos y orgánicos tienen en común el ceder iones H+ en solución acuosa, pueden presentar una disolución simple o compleja, y ser fuertes o débiles (baja disociación). Ejemplos de disociaciones:
1) Acido acético, un caso de disociación simple:
CH3COOH = CH3COO– + H+
Ka = [CH3COO–] x [H+]
Ka = 1.8 x 10-5
2) Acido carbónico, un caso de disociación compleja, donde tendremos dos constantes de disociación:
a) H2CO3= H++ HCO3–
K1 = [H+] x [HCO3–]
K1 = 10-6.4
b)HCO3–= H+ + CO3-2
K2 = [H+] x [CO3-2]
K2 = 10-10..3
como podemos observar, la primera disociación es mucho más importante que la segunda (~ 4 ordenes de magnitud) con lo cual podemos deducir que a partir de H2CO3 la proporción de iones HCO3– será muy superior a la de CO3-2 en la solución.
El agua también está sujeta a los fenómenos de equilibrio químico:
H2O = H+ + 2 OH–
Kw = [H+] x [OH–]2
Kw = 10-14
de ahí que en el agua neutra [H+] = 10-7 y el pH (-log [H+]) sea igual a 7.
1.4.- Hidrólisis
Etimológicamente la hidrólisis no es más que la descomposición de una substancia por la acción del agua, y el término proviene de un período antiguo de la química, en el que se pensaba que el agua podía dividir una sal en un ácido y una base, por ejemplo:
“CaCO3 + 2 H2O → Ca(OH)2 + H2CO3”
La realidad es diferente, más compleja, y lo que muestra son equilibrios y reequilibrios a medida que el carbonato de calcio reacciona con los hidrogeniones (H+) del agua:
CaCO3 + H+ = Ca2+ + HCO3– (a)
La reacción implica un consumo de hidrogeniones, lo cual lleva a que se ionice más agua para mantener el equilibrio:
H2O = H+ + OH– (b)
Si combinamos las reacciones (a) y (b) tendremos:
CaCO3 + H2O = Ca2+ + OH– + HCO3–
la cual representa de manera más adecuada el equilibrio que se producen, pudiéndose observar el carácter alcalino que adopta el sistema.
1.5.- Oxidación – reducción, diagramas Eh-pH
La oxidación es un cambio químico en el que un átomo o grupo de átomos pierde electrones, mientras que la reducción es el proceso mediante el cual los ganan. Así, la transformación que convierte un átomo neutro en un ión positivo es un proceso oxidativo y va acompañado por una cesión de electrones, por ejemplo:
Fe → Fe2+ + 2e–
Por el contrario, si un átomo gana electrones estamos en presencia de un proceso reductor, por ejemplo:
Cl2 + 2e– → 2 Cl–
El estado de oxidación de un átomo en un compuesto químico, es la carga eléctrica asignada a dicho átomo de manera que el conjunto sume cero. Por ejemplo el estado de oxidación del cobre en CuCl2 será de 2 (2+), y cada cloro presentará una carga negativa (1-). Podemos deducir estas cargas a partir de las propiedades periódicas de cada elemento, las cuales a su vez están relacionadas con su envolvente de electrones. Por ejemplo los elementos del Grupo I presentan en sus compuestos o en su estado iónico una carga positiva.
Los procesos de oxidación-reducción pueden ser complejos, así por ejemplo en el caso de una solución con:
Zn + Cu2+ → Zn2+ + Cu
diremos que el zinc ha sufrido una oxidación (Zn0 → Zn2+) y el cobre una reducción (Cu2+ → Cu0). Esta reacción la podemos medir mediante un dispositivo: sumergimos piezas de ambos metales en una solución de sulfato de cobre, el zinc se disolverá progresivamente, mientras que los iones de cobre de la solución se desplazarán a la pieza de cobre metal. Esto genera una corriente eléctrica medible en voltios (v). Podemos representar el proceso a través de dos semireacciones:
Zn0 → Zn2+ + 2e–
Cu2+ + 2e– → Cu0
Para darle a esta información un carácter cuantitativo, es conveniente asignarle a cada semireacción una diferencia de potencial. Esto se consigue de manera arbitraria asignando un potencial cero que denominaremos potencial estándar (E0). Una elección conveniente a este efecto es el hidrógeno:
0.5 H2 → H+ + e– E0 = 0.00 v
Si preparamos una celda con zinc como uno de los electrodos e hidrógeno en el otro (hidrógeno a 1 atmósfera burbujeando sobre un alambre de platino) y utilizamos una solución que contenga 1M de H+ y 1M de Zn2+ obtendremos:
Zn + 2 H+ → Zn2+ + H2 E0 = 0.76 v
Este valor nos dará el potencial estándar para la semireacción:
Zn → Zn2+ + 2e– E0 = 0.76 v
Estos valores se encuentran en tablas, razón por la que no hay que calcularlos para cada par.
Con el objeto de poder aplicar estos conceptos al estudio de los procesos geoquímicos que ocurren en la naturaleza debemos incluir otro término, el potencial redox. Sabemos por ejemplo que el azufre disuelto en los mares abiertos (oxigenados) se encuentra en la forma de SO42-, mientras que en charcas anóxicas, éste se encuentra bajo la forma de H2S. Esta capacidad de los ambientes naturales para oxidar o reducir compuestos es medida cuantitativamente mediante el potencial redox (Eh).
El potencial redox es medible en la naturaleza, y lo importante es que podemos unirlo al concepto de pH, de una manera tal que nos permita obtener una visión de conjunto de la estabilidad de las diferentes especies químicas a distintas concentraciones, pH, y condiciones redox. A partir de la ecuación de Nernst, y para una reacción a 25ºC en la que intervienen, A, B, C, D, podemos llegar a la expresión:
![]()
donde n es el número de electrones que intervienen en la reacción. Si analizamos por ejemplo el caso de hierro podremos construir el siguiente sistema (Fig. 1):
Fe2+ = Fe3+ + e– E0 = 0.77 v
esta ecuación es independiente del pH, de tal manera que sobre la recta de pendiente 0 cualquier razón [Fe3+]/[Fe2+] > 1. La recta sin embargo no puede ser trazada indefinidamente hacia la derecha, ya que dependiendo de la concentración de hierro, éste empezará a precipitar como Fe(OH)3 alrededor de pH 3. De esta manera necesitamos de otra reacción, en medio ácido:
(a) 3 H2O + Fe2+ = Fe(OH)3 + 3 H+ E0 = 0.98 v
pero, nuevamente, a medida que progresamos hacia la izquierda de nuestro diagrama de Eh-pH, empezamos entrar progresivamente en condiciones básicas (alcalinas), de tal manera que necesitamos otra reacción que denote adecuadamente dichas condiciones:
(b) Fe(OH)2 + OH– = Fe(OH)3 + e– E0 = -0.56 v
¿Cómo conectamos estas reacciones con el pH? a partir de (a) tenemos:
![]()
para [Fe2+] = 1M, E0 = 0.98 v, y dado que n = 1, entonces:
Eh = 0.98 + 0.059 log [H+]3 – 0.059 log 1
Eh = 0.98 + 0.177 [H+]
dado que pH = -log [H+], entonces:
Eh = 0.98 – 0.177 pH
y para una concentración de [Fe2+] = 10-7M tendremos:
Eh = 0.98 + 0.059 log [H+]3 – 0.059 log -7
Eh = 1.40 – 0.177 pH
En el caso de condiciones básicas (b) y una concentración de [Fe2+] = 1M tendremos:
![]()
dado que n = 1, y el potencial estándar es de -0.56, entonces:
![]()
dado que [H+] x [OH–] = 10-14 (Kw), despejando tenemos:
Eh = 0.27 – 0.059 pH
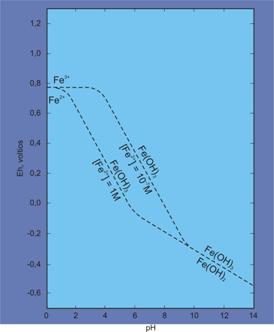
Diagrama Eh-Ph para el hierro a concentraciones de Fe2+ de 1M y 10-7M
Estas ecuaciones nos permiten evaluar las condiciones de estabilidad del hierro en los distintos ambientes de pH y Eh. Dado que el hierro migra como Fe2+ y precipita como Fe3+(Fe(OH)3), bastará con que sepamos cual es la concentración de hierro, para poder determinar la estabilidad en solución del catión ferroso. Esta metodología la podemos utilizar para metales individuales o para comparar el posible comportamiento de pares catiónicos, por ejemplo, hierro y manganeso en solución como especies reducidas (Fe2+, Mn2+).
1.6.- Bibliografía
Gill, R. 1996. Chemical fundamentals of geology. Chapman & Hall, London, 290 pp.
Krauskopf, K.B. 1979. Introduction to geochemistry. McGraw-Hill, NY, 617 pp.
Petrucci, R.H., y Harwood, W.S. 1993. General chemistry: principles and modern applications. MacMillan, NY, 1000 pp.
Rosemberg, J.L. 1969. Química general. McGraw-Hill, NY, 246 pp.
2.- Solubilización, transporte, y precipitación de…
Solubilización, transporte, y precipitación de substancias en el ciclo exógeno: una aproximación al tema de los contaminantes
Higueras1y R. Oyarzun2
1: Departamento de Ingeniería Geológica y Minera, Escuela Universitaria Politécnica de Almadén, Universidad de Castilla-La Mancha, Plaza M. Meca 1, 13400 Almadén, España.
2: Departamento de Cristalografía y Mineralogía, Facultad de Ciencias Geológicas, Universidad Complutense, 28040 Madrid, España.

Nacimiento del río Cuervo (Cuenca)
A pesar de que existen diferentes vías de movilización de las llamadas substancias contaminantes, el medio acuático tiene una particular relevancia, ya que éste permite el transporte por largas distancias de innumerables compuestos, y comprende ambientes muy variados. Dado que el agua sustenta la vida del planeta, sus características químicas son de la mayor importancia. Cuando hablamos del medio acuático nos referimos a las aguas subterráneas, ríos, lagos, y mares y océanos. La relación del hombre con el medio acuático ha estado siempre presente, en sus orígenes como fuente para beber, y más tarde para regar los cultivos, como medio de comunicación, y finalmente para su utilización en procesos industriales.

Navegación en el río Rin, Alemania.
De esta manera el hombre siempre se ha establecido cerca de los medios acuáticos, sirviéndose directamente de ellos de distintas maneras. Primero fueron pequeñas aldeas en la rivera de los ríos, luego comunidades mayores, y finalmente las modernas ciudades que continuaron esta relación con los cursos fluviales.

La ciudad de Paris y el río Sena.
Lo mismo ocurrió en las zonas costeras, en el entorno de lagos o mares. El problema ambiental surge del hecho de que el hombre también utiliza directa o indirectamente este medio como destino final de sus residuos, urbanos o industriales.

Vertidos en un curso de agua.
Hasta hace pocas décadas atrás existían escasos reglamentos que rigieran dichas actividades, y mientras más desarrollado el país o región del mundo, más substancias peligrosas eran vertidas a los cursos fluviales, lagos y océanos, o infiltradas a las aguas subterráneas. La explicación es simple: el desarrollo conlleva la industrialización, y ésta, los residuos. En los países de la Unión Europea el vertido de residuos (sólidos o líquidos) está fuertemente regulado en la actualidad, lo cual no significa necesariamente que no existan vastas zonas del continente que presentan serios problemas de contaminación por metales pesados u otras substancias tóxicas.
Este es un tema muy serio, en el cual debemos ser cautos, y utilizar el método científico antes que dejarnos llevar por interpretaciones a la ligera. Por ejemplo, solemos pensar en el hombre como el mayor agente contaminador del planeta, olvidando así un hecho muy importante: la Tierra es ante todo un complejo sistema físico-químico y biológico, en el cual en todo momento se están produciendo reacciones químicas de origen inorgánico u orgánico, y claro está, se trata de un sistema que aporta y recicla compuestos químicos. Nuestro planeta es un sistema en perpetuo cambio, y esto ha sido así desde sus comienzos, hace unos 4500 millones de años. Por ejemplo, hoy en día se discute en las más diversas instancias sobre el llamado cambio climático global, dándose a entender, directa o indirectamente que la industria moderna sería la gran causante de éste. Al respecto sería importante indicar que la idea de cambio climático global es largamente conocida para los que trabajan en el campo de la geología, ya que ni los continentes ni el clima han permanecido estables jamás a lo largo de la historia geológica de la Tierra. Obviamente no es nuestra intención aquí entrar en esta polémica, aunque por otra parte, resulta ineludible intentar precisar qué fenómenos pueden ser directamente achacables al hombre (procesos antropogénicos), y cuales, al curso normal de los procesos geológicos del planeta (procesos naturales). Por ejemplo, aun asumiendo que el CO2 fuera un importante gas de efecto invernadero (a pesar de la creencia general, esta es una idea contendida: ejemplo 1 – ejemplo 2), habría que precisar, entre otras cosas, cuanto del CO2 presente en la atmósfera es aportado naturalmente (entre otros) por la actividad volcánica. En efecto, no debemos olvidar que los volcanes no solo expulsan lava o ceniza, sino que también aportan grandes cantidades de gases a la atmósfera, por ejemplo, CO2 y SO2.

Imagen en falso color (NASA) mostrando las emisiones de SO2 (rojo) desde el volcán Etna (Sicilia, Italia), el 29 de Julio de 2001.
Más aun, la principal cadena volcánica del planeta, esto es, el sistema de dorsales oceánicas (que limita las distintas placas oceánicas), permanece oculta a nuestra vista bajo la superficie de los océanos, y no sabemos realmente cuanto CO2 (u otros gases) aporta al sistema atmosférico.
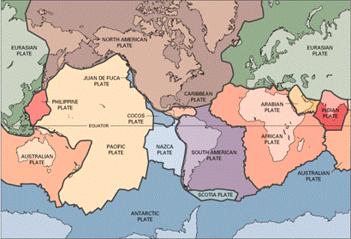
Parte del sistema de dorsales oceánicas (líneas negras gruesas) y placas (USGS).

Actividad hidrotermal submarina en la dorsal de Juan de Fuca (ver figura anterior). Observe la introducción de compuestos químicos sulfurados (penacho negro).
Por otra parte, la degasificación del planeta a partir de las áreas volcánicas no sucede solamente durante las erupciones, sino que permanece en funcionamiento durante toda la vida activa de un volcán.

Emanación de gases desde fumarolas en un área volcánica.
Finalmente, la actividad volcánica no es el único mecanismo geológico de degasificación de CO2 conocido, existiendo otros procesos más complejos que no serán discutidos aquí (e.g. degasifición a partir de complejos metamórficos).
Así, intentaremos en este capítulo revisar de una manera simple cómo se regulan físico-químicamente los sistemas geológicos exógenos, para poder entender en lo posible cómo y porqué las substancias químicas ingresan en el medio acuático, como migran dentro de él, y cual es su destino final.
Organización del Capítulo:
2.1.- Límites de Eh-pH en la naturaleza
2.2.- ¿Cómo acceden las substancias químicas al medio acuático?
2.3.- Transporte e inmovilización de metales y fases minerales en el medio acuático
2.4.- Suelos: la vulnerable piel del planeta
2.5.- Bibliografía
2.1.- Límites de Eh-pH en la naturaleza
Para que la geoquímica constituya una herramienta útil de verdad, debe tener un carácter predictivo, y para que esto ocurra debemos conocer, al menos de una manera general, las condiciones de Eh-pH que encontrarán las substancias solubles en el medio natural.
Las soluciones más ácidas que podemos encontrar en la naturaleza se encuentran cerca de centros volcánicos donde se disuelven gases de origen magmático, y cerca de masas de sulfuros sujetos a procesos de oxidación. En dichos lugares las soluciones pueden alcanzar valores de pH menores a 0 ([H+] > 1 M). Sin embargo, estos valores no perduran demasiado en el espacio ya que las soluciones reaccionan con las rocas del entorno (proceso de hidrólisis), generando un aumento del pH por consumo de hidrogeniones. Por ejemplo, analicemos la hidrólisis de un feldespato potásico, constituyente normal en una roca ígnea. El primer paso nos llevará a la formación de un mineral tipo mica potásica:
3 KAlSi3O8 + 2 H+ → KAl3Si3O10(OH)2 + 6 SiO2 + 2 K+
pero si hay abundancia de hidrogeniones (medio extraordinariamente ácido), esta fase no será estable, hidrolizándose a su vez, y dando como producto una fase mineral tipo arcilla caolinítica:
2 KAl3Si3O10(OH)2 + 2 H+ + 3 H2O → 3 Al2Si2O5(OH)4 + 2 K+
Reconoceremos este proceso en el campo por un “blanqueo” muy conspicuo de las rocas. Si además hay presentes especies oxidadas de hierro en el sistema (producto de la oxidación de sulfuros), las rocas adquirirán colores blancos y rojizos (agregado de arcillas caoliníticas y limonitas).

Rocas piroclásticas alteradas a arcillas, con recubrimiento de limonitas.
Así, debido a los procesos de hidrólisis que se producen al interaccionar las soluciones ácidas con las fases minerales reactivas de las rocas, resulta difícil que perduren las condiciones extremas de bajo pH. De cualquier manera, nunca se producirá una neutralización total de las soluciones, ya que no debemos olvidar que el CO2 atmosférico reacciona con el agua formando ácido carbónico (H2CO3). Por otra parte el decaimiento de la materia orgánica da origen a ácidos orgánicos. En cualquier caso los valores de pH que se obtengan por estos procesos no serán nunca comparables con aquellos antes discutidos. Así lo típico es que las aguas superficiales tengan valores de pH en el orden de 5 a 6. Valores de pH más bajos pueden no obstante encontrase en los horizontes A de suelos tipo pedalfer o podzol donde se puede llegar a un pH de 3.5. De esta manera, y obviando los valores extremos, podemos fijar un límite inferior de pH en torno a 4 en los ambientes naturales. En el otro extremo de la escala podemos encontrar valores de 10 en aguas libres de CO2 que reaccionan con rocas carbonatadas. En cuencas salinas de regiones desérticas los valores de pH pueden llegar incluso a 12. Nuevamente, olvidándonos por un momento de estos valores extremos, se puede fijar un límite superior razonable en 9.
En lo que se refiere a fenómenos redox, digamos que el agente natural más oxidante en la naturaleza es precisamente el oxígeno. Así el límite superior del potencial redox viene dado por la reacción:
H2O = 0.5 O2 + 2 H+ + 2 e– E0 = 1.23 V
El potencial de esta reacción depende del pH:
Eh = 1.23 + 0.03 log [O2]0.5[H+]
Como concentración normal de oxígeno podemos utilizar el valor de 0.2 atm (el oxígeno ocupa un quinto en volumen del aire que respiramos), así tendremos:
Eh = 1.23 + 0.03 log (0.2) 0.5 + 0.059 log [H+]
Eh = 1.22 – 0.059 pH
En el extremo reductor de los procesos redox nos encontramos con el hidrógeno a través de la reacción:
H2 = 2 H+ + 2 e– E0 = 0.00 V
para la cual tendremos:
Eh = 0.00 + 0.03 log [H+]2 – 0.03 log [H2]
Dado que la concentración de hidrógeno en ambientes naturales cercanos a la superficie no puede superar a 1 atm tendremos:
Eh = 0.00 + 0.03 log [H+]2 – log 1
Eh = – 0.059 pH
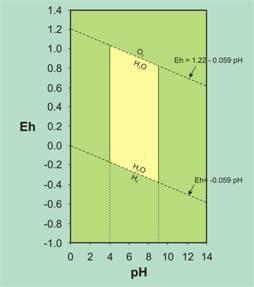
Límites de los valores normales de Eh-pH de las aguas (en amarillo).
Conviene aquí insistir que el cuadro que emerge de estas disquisiciones comprende “la mayoría de los ambientes naturales” pero no “todos”. Como hemos visto, existen ambientes de mayor acidez o alcalinidad que no quedan reflejados en la figura anterior.
2.2.- ¿Cómo acceden las substancias químicas al medio acuático?
Existen tres vías principales por las cuales las substancias químicas de origen natural o antropogénico acceden al medio acuático: 1) mediante el traspaso atmósfera-hidrósfera (por absorción del gas en la fase líquida); 2) mediante la solubilización de substancias sólidas; y 3) mediante el vertido directo de líquidos.

Drenaje ácido procedente de la mina de San Quintín (Ciudad Real, España)
2.2.1.- Traspaso atmósfera-hidrósfera
2.2.2.- Solubilización de substancias sólidas
2.2.3.- Vertido directo de líquidos
2.2.1.- Traspaso atmósfera-hidrósfera
En el caso del traspaso atmósfera-hidrósfera debemos clarificar en primer lugar la fuente del gas en cuestión. Analizaremos dos casos principales (CO2 y SO2), y de manera breve un tercero (Hg). El CO2 y SO2 pueden incorporarse a la atmósfera por procesos naturales o antropogénicos. En el caso del CO2 debemos a su vez diferenciar entre los procesos geológicos y los biológicos. Como se explicaba en párrafos anteriores, la actividad volcánica suministra gases a la atmósfera, siendo el CO2 uno de los principales. Esta degasificación es continua, esto es, no se restringe a los episodios de actividad eruptiva de un volcán, ni al cono en particular. De hecho los cinturones volcánicos pueden considerarse como amplias zonas de degasificación de aporte continuo, siendo el ejemplo más ilustrativo la actividad fumarólica asociada. Las zonas de geysers o de surgencias hidrotermales (hot-springs), también relacionadas con la actividad volcánica, constituyen a su vez otras fuentes de aporte.

Un geyser en actividad.
Estas aguas termales (hot-springs) contienen generalmente CO2 disuelto, que llegado el momento es transferido a la atmósfera de una forma equivalente a lo que ocurre con una bebida gasificada cuando es abierta. La interacción de las soluciones hidrotermales con rocas carbonatadas en profundidad también libera CO2, el que asciende a la superficie a través de fracturas.
Hasta aquí de manera muy resumida lo que podríamos denominar “algunas” fuentes geológicas de aporte de CO2 a la atmósfera. Pero los procesos naturales de aporte no acaban en la actividad geológica, ya que la actividad biológica también contribuye al inventario de CO2 atmosférico. Por ejemplo, solemos pensar en las plantas como fijadoras de CO2, y esto es correcto, pero solo en parte (ecuación global de la fotosíntesis):
6 CO2 + 12 H2O → C6H12O6 + 6 O2 + 6 H2O
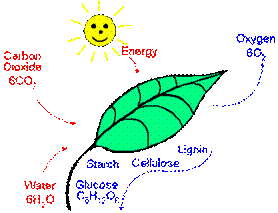
El proceso de la fotosíntesis.
Los azúcares generados así durante la fotosíntesis son empleados por la planta en su metabolismo, para su mantenimiento y crecimiento. Llegados a este punto, es necesario aclarar que el proceso funciona bien “solo” mientras la planta vive. Una vez que la planta muere, el carbono que ha utilizado en la construcción de su organismo decae a compuestos orgánicos simples (CHx). En condiciones aireadas esta materia orgánica es oxidada dando como producto CO2 (CHx → CO2). Podemos representar esta situación a través del proceso de oxidación del metano, típico producto de la descomposición de materia orgánica:
CH4 + 2 O2 → CO2 + 2 H2O
En otras palabras, al menos parte del CO2 que la planta tomó de la atmósfera durante su ciclo de vida es retornado a ésta. Otra fuente biológica de generación de CO2 es el proceso de respiración en los animales, los cuales inhalan oxígeno y expulsan dióxido de carbono.
En lo que corresponde a procesos antropogénicos, la principal fuente de CO2 proviene de la combustión de maderas y combustibles fósiles (carbón y derivados del petróleo).

Combustión de carbones para la generación de energía eléctrica.
El aporte antropogénico a la atmósfera sería de unas 24 Gt (Giga toneladas: 109 toneladas) de CO2 por año, aunque el problema consiste en estimar que proporción de éstas son fijadas por la actividad biológica (e.g. respiración y crecimiento de plantas) o geológica (e.g. formación de CaCO3). Por ejemplo, el CO2 atmosférico reacciona con el agua de lluvia formando ácido carbónico:
CO2 + H2O → H2CO3
A su vez, el ácido carbónico es un potente disolvente de los carbonatos, y una prueba de ello es la formación de cavernas en terrenos carbonatados:
CaCO3 + H2CO3 → Ca2+ + 2 HCO3–

Formación de cuevas por disolución de material carbonatado.
Así, tanto en los medios terrestres como acuáticos, la cantidad de CO2 disponible rige el sistema de precipitación – disolución de los carbonatos:
CO2 + H2O
↨
CaCO3 + H2CO3 ↔ Ca2+ + 2 HCO3–
Mientras más dióxido de carbono esté disponible habrá una mayor formación de ácido carbónico, y a una mayor cantidad de ácido carbónico generará más disolución de carbonato de calcio. Lo mismo rige a la inversa en los mares cálidos. ¿Cómo es esto así? imaginemos un refresco de cola: si abrimos la botella cuando el líquido está tibio generaremos una salida explosiva de CO2, ya que su solubilidad es dependiente de la temperatura. De la misma manera, en los mares cálidos el CO2 es menos soluble, escapando a la atmósfera, lo que hace que haya menos H2CO3disponible, y por lo tanto se favorezca la precipitación inorgánica y orgánica de carbonatos. ¿Cómo podemos comprobar que esto ocurre así efectivamente? fácil, observe donde se encuentran las formaciones coralinas más importantes del mundo. En presencia de mayores concentraciones de ácido carbónico (mares fríos) los esqueletos calcáreos de los corales no son viables.
Aun hoy no existe un consenso con respecto a que está ocurriendo efectivamente con el CO2 a escala planetaria. Sabemos que en los últimos siglos ha habido un incremento en la concentración de CO2 atmosférico, desde unas 280 ppm (en la época pre-industrial) a las 360 ppm actuales. Desgraciadamente no se cuenta con datos fiables más allá de 1960, por lo que resulta difícil explicar “completamente” un fenómeno que “podría” estar ligado (efectivamente) al incremento en las emisiones industriales de CO2. Este es un tema en el cual toda cautela es poca ya que no debemos olvidar, que al igual que el clima, las concentraciones de CO2 en la atmósfera han variado sensiblemente a lo largo de los tiempos geológicos, sin que mediara, obviamente para nada, la intervención del hombre. Pero más aun ¿ que pasa si el CO2 después de todo no tiene que ver directamente con el calentamiento global o alternativamente, que sí está relacionado, pero las relaciones causa – efecto son a la inversa ? Al igual que en medicina, antes que aplicar un tratamiento, es importante diagnosticar adecuadamente la dolencia, para evitar en lo posible confundir los síntomas con la enfermedad (si tomamos aspirinas bajamos la fiebre pero no curamos la infección…).
Otro peligro potencial derivado de las emisiones de CO2 al medio acuático proviene de una fuente volcánica, esto es, la degasifición de una cámara magmática somera bajo un lago. El peligro de tal situación quedó de manifiesto en la tragedia del lago Nyos (1986), en Camerún.

El lago Nyos (Camerún).
A lo largo de Camerún pasa la Línea Volcánica del mismo nombre, una zona de vulcanismo activo que se extiende desde el Atlántico hacia Africa según una dirección NE. En sitios protegidos, como por ejemplo una caldera volcánica, un lago puede sufrir el fenómeno de “estratificación”, es decir, la columna de agua presenta una diferenciación en capas debido variaciones composicionales (sales o gases disueltos) o térmicas. Así la capa superior, de mayor temperatura “flota” sobre otra profunda de menor temperatura y mayor densidad. En las regiones temperadas esta estratificación se rompe naturalmente cuando el enfriamiento estacional permite la mezcla entre ambas capas (overturn: aquí expresado como “inversión”). Sin embargo, en regiones tropicales, con temperaturas más o menos estables durante todo el año, la inversión resulta más difícil. Sin embargo, si una capa profunda con gases disueltos recibe un aporte sostenido de los mismos, estos acabarán por emerger a la superficie de manera catastrófica una vez superado el nivel de saturación de las aguas. Este es muy probablemente el caso del lago Nyos, donde el 21 de Agosto de 1986 emergió una gigantesca “burbuja” de dióxido de carbono (~ 109 m3 CO2), que durante su avance por valles y desniveles (el CO2 es más 1.5 veces más pesado que el aire) mató a miles de personas y animales por asfixia.
Finalmente, señalemos que el CO2 también cumple un papel extraordinariamente positivo en las aguas en lo que se refiere al control del pH. Si recordamos lo estudiado anteriormente en el capítulo, el CO2 reacciona con el agua formando ácido carbónico. Este último a su vez se disocia formado bicarbonato y carbonato:
H2CO3 = H+ + HCO3–
HCO3– = H+ + CO32-
Si introducimos hidrogeniones en el agua, tanto el ión bicarbonato (HCO3–) como el carbonato (CO32-) reaccionarán con éstos, manteniendo de esta forma relativamente estable el pH del medio acuoso (se introducen H+ pero estos son consumidos, manteniéndose el equilibrio: buffering):
H+ + HCO3– = H2O + CO2
2 H+ + CO32- = H2O + CO2
El caso de los gases oxidados de azufre (SO2, SO3) presenta similitudes con el dióxido de carbono en lo que se refiere a las fuentes de origen geológico. Determinados tipos de vulcanismo (e.g. el adakítico) son muy ricos en especies oxidadas de azufre (sistemas de alta fO2). Por ejemplo, erupciones relativamente recientes como las del Chichón en México (1982) o la del Pinatubo en Filipinas (1991) generaron cantidades de 7 y 20 Mt (Mega toneladas: 106 toneladas) de SO2 respectivamente. El sistema en estos casos es tan rico en sulfato que da origen a la formación directa (primaria) de sulfato de calcio (anhidrita: CaSO4), en las rocas volcánicas formadas durante el proceso eruptivo.
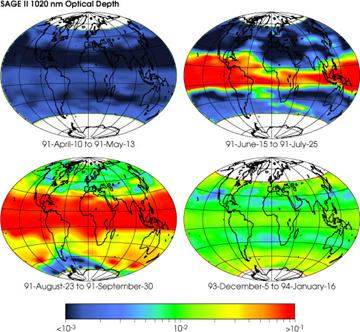
Evolución en el tiempo del SO2 inyectado a la atmósfera por el volcán Pinatubo, en relación a la erupción del año 1991 (NASA).
De cualquier manera, las fuentes de SO2 de origen antropogénico son muy importantes, y se derivan principalmente de dos fuentes: la combustión de carbones con pirita y la fundición de sulfuros para la extracción de metales en las instalaciones mineras (pirometalurgia). En ambos casos estamos tratando con sulfuros como fuente primaria de SO2. Los carbones tienen contenidos variables de pirita (FeS2), y lo que ocurre en las centrales eléctricas alimentadas por carbón es el siguiente proceso:
4 FeS2 + 11 O2 → 8 SO2 + 2 Fe2O3
lo que en esencia es muy parecido a las reacciones principales que ocurren dentro de un horno convertidor en la metalurgia del cobre (producción de cobre blister):
2 Cu2S + 3 O2 → 2 Cu2O + 2 SO2
2 Cu2O + Cu2S → 6 Cu + SO2

La refinería de cobre de Chuquicamata, Chile.

Convertidores para la fundición de sulfuros y producción de cobre metálico.
Para que nos hagamos una idea de cuanto SO2 pueden expulsar anualmente las chimeneas de una gran refinería de cobre, como la de Chuquicamata (ver foto arriba), comentemos que en el año 1999 dichas instalaciones arrojaron 255.000 t de este contaminante. Todas las reacciones de arriba expresadas producen SO2, el cual en contacto con las gotas de agua en la atmósfera reacciona produciendo finalmente ácido sulfúrico:
SO2 + H2O → H+ + HSO–
2 HSO– + O2 → 2 H+ + 2 SO42-
Este ácido sulfúrico es el causante de la denominada lluvia ácida, la cual en ausencia de agentes reguladores (buffers) es capaz de bajar el pH de las aguas de lagos y ríos, causando la muerte de la vegetación y de la fauna acuática.

Daños en la vegetación inducidos por la lluvia ácida.
No solo los carbones contienen azufre (0.2-7 % S), sino que otros combustibles también son ricos en este elemento, por ejemplo el fuel (0.5-4 % S), el diesel (0.3-0.9 % S), la gasolina (0.1 % S), y el keroseno (0.1 % S). La madera y el gas natural presentan muy bajos contenidos en azufre, aunque no olvidemos que el proceso de combustión es generador de CO2.
En la metalurgia del cobre la medida paliativa más importante consiste en la producción de ácido sulfúrico a partir del SO2 producido. Los pasos son los siguientes: 1) conversión del SO2gaseoso a SO3, y 2) conversión subsiguiente del SO3 en ácido sulfúrico:
SO2 + 0.5 O2 → SO3 (1)
SO3 + H2 → H2SO4 (2)
Esta medida, de amplia utilización en países mineros tales como USA, Canadá, o Chile ha tenido consecuencias inesperadas. Por ejemplo, en Chile se generó un exceso de ácido sulfúrico al que hubo que buscarle una salida económica y ambiental. Esta llegó de mano de la hidrometalurgia, la cual utiliza justamente ácido sulfúrico diluido, para lixiviar cobre (lixiviación en pila: heap leaching) a partir de oxidados de este metal, e incluso sulfuros.

Operación de lixiviado en pila en la localidad de Panulcillo (Chile).
Hoy en día en Chile una parte importante del cobre obtenido en las operaciones mineras proviene de procesos de este tipo. Dado que el cobre lixiviado es capturado por solventes orgánicos (extracción por solventes), y finalmente extraído por electrorecuperación (un proceso conocido internacionalmente como SX-EW), la pirometalurgia no es necesaria. Así, una industria potencialmente contaminante da pie a otra con una tecnología más limpia.
Otras emisiones que pueden tener muy graves consecuencias son las relacionadas con el mercurio. El mercurio puede ser incorporado a la atmósfera a través de procesos naturales (actividad volcánica), y la actividad minera e industrial.
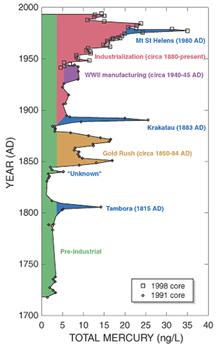
Fuentes de emisión de mercurio a la atmósfera. Note (en azul) las emisiones de los volcanes Tambora, Krakatoa, y Santa Elena (USGS).
En primer lugar está la metalurgia en sí de las menas mercuriales (cinabrio: HgS), cuya metalurgia genera importantes emisiones de mercurio gaseoso a la atmósfera. Por ejemplo, en la metalurgia de Almadén (España), cerca de las instalaciones del complejo metalúrgico, se han detectado valores de alrededor de 15000 ng Hg m-3, una cifra altísima considerando que dichas instalaciones están dentro del pueblo de Almadén, y que los valores máximos admitidos por la Organización Mundial de Salud son de 1000 ng Hg m-3.

Parte del complejo metalúrgico de Almadén (España).
La segunda fuente más importante de mercurio es la amalgamación de oro en pequeñas operaciones mineras, generalmente localizadas en los llamados países del tercer mundo. Una vez que el oro ha sido amalgamado, se intenta recuperar el máximo posible de mercurio, aunque una parte permanece unida al oro. Por esta razón los mineros calientan la amalgama en una placa, enviado así ese mercurio a la atmósfera. Sin embargo ese mercurio gaseoso puede retornar al suelo, ya sea como precipitación directa de Hg2+ o por conversión de Hg0 a Hg2+, mediante procesos mediados por el ozono, u otros (g: fase gas; aq: fase acuosa; p: fase particulada):
Hg0 (g) → Hg0(aq)
Hg0(aq) + O3(aq) → Hg2+(aq)
Hg2+(aq) + hollín/posible evaporación → Hg2+(p)
Aunque en los suelos existe una tendencia a la reducción del ión Hg2+ a Hg0(g) (retorno a la atmósfera) mediante procesos fotolíticos, parte de este mercurio puede incorporarse a las aguas. De cualquier manera, los riesgos más importantes de contaminación de aguas por mercurio no provienen de la industria minera, sino que de otras actividades industriales o de silvicultura, que utilizan compuestos clorurados de mercurio. Por ejemplo el HgCl2 es utilizado como desinfectante, fungicida, insecticida, y preservante de la madera, en tanto que el Hg2Cl2 es empleado en la industria farmacéutica y en electrodos. Y el problema, aunque parezca una paradoja, no radica tanto en estos compuestos (que per se son peligrosos), sino en la posibilidad de que se forme metil mercurio (CH3Hg+) a partir de las especies cloruradas. El metil mercurio pasa a través de la cadena trófica a la fauna mayor (peces) y de ahí al ser humano, donde puede ocasionar trastornos de salud extremadamente graves.
2.2.2.- Solubilización de substancias sólidas
Uno de los problemas más serios de introducción de metales pesados en el medio acuoso viene dado por la disolución de especies minerales que están en desequilibrio con las condiciones del medio que las contiene, por ejemplo, un sulfuro metálico en condiciones oxidantes. Este caso es particularmente importante en regiones del mundo donde existe o ha existido una importante industria minera. ¿Por qué centrar esta discusión en la minería? porque la minería constituye la fuente más importante metales en el mundo (una parte pequeña proviene de operaciones de reciclado). La minería, como cualquier otro proceso industrial, produce problemas en la calidad del agua en dos aspectos principales: 1) desarrollo del llamado drenaje ácido de mina como consecuencia de la oxidación y lixiviación (solubilización) de las especies sulfuradas, lo que acarrea un descenso del pH y la contaminación de aguas, debido a la propia naturaleza de los materiales explotados, por ejemplo metales pesados tales como Cu, Pb, Zn-(Cd), As, Hg, etc. y aniones asociados (sulfatos, carbonatos, etc.); y 2) contaminación de las aguas debido al uso de técnicas de lixiviación en pila (heap leaching) de metales, donde el agente lixiviante puede ser el ácido sulfúrico (para el cobre) o el cianuro de sodio (para el oro). Analizaremos el primer caso en esta sección, y el segundo en la siguiente.
Sin duda, el mayor problema que representa la minería frente a las aguas es la formación del denominado drenaje ácido de mina, consistente en la formación de aguas de gran acidez, por lo general ricas en sulfatos, y con contenidos variables en metales pesados. Dicho drenaje se desarrolla a partir de la lixiviación de sulfuros metálicos o de pirita presente en carbones. Para ello existen dos fuentes principales: 1) el mineral sulfurado in situ (causa no antropogénica), y 2) las escombreras de minerales (mineral dumps) y balsas de estériles (tailings) abandonadas.

Drenaje ácido de mina. Note la fuerte coloración rojiza, dada por especies oxidadas de hierro.
No obstante, en algunos casos los fenómenos naturales, ejemplificados en el caso del Río Tinto (Huelva), pueden llegar a alcanzar grandes proporciones. El drenaje ácido se produce por la oxidación e hidrólisis de los sulfuros, y en especial de la pirita, mediante la serie de reacciones:
4 FeS2 + 14 O2 + 4 H2O → 4 Fe2+ + 8 SO42- + 8 H+
a su vez, los iones ferroso (Fe2+) se oxidarán de la siguiente manera:
4 Fe2+ + O2 + 4 H+ → 4 Fe3+ + 2 H2O
los iones férricos se hidrolizan para formar goethita:
Fe3+ + 2 H2O → 4 FeO(OH) + 3 H+

Procesos naturales de oxidación de sulfuros (principalmente pirita) en el yacimiento de Río Tinto (Huelva. España). El color rojizo se deriva de la presencia masiva de goethita.
En climas muy áridos (Desierto de Atacama, Chile; Outback de Australia), esta serie de reacciones puede quedar interrumpida dando origen a sulfatos férricos tales como jarosita, copiapita o coquimbita.
A pesar de que estas reacciones pueden dar a entender que suceden en condiciones puramente inorgánicas, el entorno biológico juega un papel decisivo. Por ejemplo, la bacteria Thiobacillus ferrooxidans es la mayor responsable de la contaminación relacionada con el drenaje ácido procedente de explotaciones mineras y mineralizaciones en general. Esta es una bacteria acidófila (propia de ambiente ácido), con una fisiología basada en la fijación de carbono a partir del CO2 atmosférico, obteniendo su energía a partir de oxidación del hierro (1) o azufre (2):
Fe2+ + 0.25 O2 + H+ → Fe3+ + 0.5 H2O (1)
H2S + 2 O2 → SO42- + 2 H+ (2)
En ausencia de bacterias del tipo T. ferrooxidans, las reacciones de oxidación de los sulfuros se ven severamente ralentizadas.
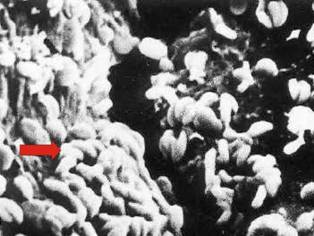
Bacterias T. ferrooxidans (flecha) adheridas a una partícula de sulfuro.
Otro factor que incide de manera decisiva en la oxidación y lixiviación de sulfuros metálicos es la tasa de producción de sulfato férrico:
Fe2(SO4)3 = 2 Fe3+ + 3 SO42-
el cual actúa como un potente agente oxidante para minerales tales como la pirita (1) o calcopirita (2):
FeS2 + 14 Fe3+ 8 H2O → 15 Fe2+ + 2 SO42- + 16 H+ (1)
CuFeS2 + 16Fe3+ + 8 H2O → Cu2+ + 2 SO42- + 17 Fe2+ (2)
El problema ambiental derivado de estos procesos radica no solo en la liberación de cationes relativamente inocuos como Fe2+ o Fe3+, o en que aumente la acidez de las aguas. Aunque estos son dos problemas en sí, nos enfrentamos además a otro grave peligro para la salud humana y ambiental: el resto de los minerales sulfurados presentes en una escombrera o balsa de estériles también se oxidan, liberando metales pesados de altísima peligrosidad tales como (entre otros) el plomo (presente en galena, 1), el arsénico (presente en arsenopirita, 2 o enargita, 3), o el cadmio (presente en la fase de esfalerita, 4):
2 PbS + 4 Fe3+ +3 O2 + 2 H2O → 2 PbSO4 + 4 Fe2+ + 4 H+ (1)
2 FeAsS + Fe2(SO4)3 → 2 H3AsO4 + 4 FeSO4 + H2SO4 (2)
Cu3AsS4 + 5.5 Fe2(SO4)3 + 4 H2O → 3 CuSO4 + 11 FeSO4 + 4 S + H3AsO4 + 2.5 H2SO4 (3)
Zn1-xCdxS + 8 Fe3+ + 4 H2O → 8 H+ + (1-x) Zn2+ + (x) Cd2+ + SO42- + 14 Fe2+ (4)
Así, nos enfrentamos primariamente a un problema de “mineralogía ambiental”, y en segundo lugar, a otro de “geoquímica ambiental”. En ocasiones la opinión pública suele fijar sus críticas sobre el impacto ambiental minero más evidente, esto es, la presencia en si de una mina (impacto visual), olvidando los aspectos mineralógicos y químicos que se derivan de la actividad como tal. Aquí hay varios temas a tratar: 1) la “mineralogía” del yacimiento que se explota; 2) el o los “metales” presentes en esas fases minerales; 3) los “procesos metalúrgicos” que se emplean para extraer el metal o metales; y 4) el “clima” de la región. Obviamente no es lo mismo bajo el punto de vista de la salud humana una explotación minera de hierro, que otra de arsénico o plomo. La primera podrá generar importantes impactos visuales o sociales, pero el hierro definitivamente no está dentro de la lista de elementos químicos de alta peligrosidad. Así, el problema debe ser enfocado primariamente en términos de la mineralogía del yacimiento que ha sido explotado o se encuentra en explotación. Quisiéramos insistir sobre este último punto. Continentes como el europeo registran una historia minera y metalúrgica que se extiende, de manera importante, desde los tiempos del Imperio Romano. Esta minería ha dejado un legado, muchas veces oculto, de escombreras de minerales que con el pasar de los siglos han sido “disimuladas” por la acción de la naturaleza o actividades humanas.

Escombreras y lodos estériles en el Distrito de la Unión (Murcia, España), sector de Trinidad, desde la cuesta de Las Lajas. Los estériles comienzan a ser cubiertos por la vegetación.
La carga mineral que no se explotó en su momento por razones de atraso tecnológico, o por las mismas, se explotó solo de manera parcial, está ahí, sujeta a procesos químicos que actúan sobre dichos minerales. Sin ir demasiado lejos en el tiempo, en España existen cientos de minas abandonadas (con sus escombreras y/o balsas asociadas, cuya explotación cesó en el siglo 19 o 20. La inmensa mayoría de estas presentan riesgos ambientales que aun no han sido evaluados o evaluados del todo. Por ejemplo, sabemos de la presencia de antiguas explotaciones de plata en la Sierra de Guadarrama (e.g. Bustarviejo), cuya mineralogía incluye entre otros, minerales de arsénico (arsenopirita). O que decir de las minas de mercurio del distrito de Almadén (Ciudad Real), o de plomo y zinc del distrito de la Unión (Murcia). Estas minas, y sus escombreras y balsas de estériles constituyen auténticas “bombas de tiempo químicas”. Urge en este sentido en España una política a nivel local (ayuntamientos), regional (gobiernos autonómicos), y nacional, que plantee el estudio de una manera “seria y competente” (más allá de la típica palabrería de costumbre) de los potenciales peligros ambientales de salud derivados de las cargas metalíferas dispersas por todo el territorio nacional.

Escombreras abandonadas y drenaje ácido en la antigua explotación de Pb-Zn de San Quintín (Ciudad Real, España).
La metalurgia empleada en su momento para extraer el o los metales de interés económico es otro de los temas de interés llegado el momento de evaluar la peligrosidad de una explotación minera. En este sentido conviene recordar los conceptos de “mena” y “ganga”. Puesto en términos muy simples, mena es el o los minerales que presentan un interés económico en un determinado momento por razones de mercado. Ganga es la fase mineral que carece de dicho interés económico. De esta manera, para una compañía puede ser interesante bajo el punto de vista económico explotar uno o dos minerales mientras considera a un tercero como ganga. El problema radica en que pasa si este último mineral contiene una fase metálica de alta peligrosidad ambiental. Por ejemplo, un mineral típico presente en los yacimientos de sulfuros masivos de la Faja Pirítica Ibérica es la arsenopirita (FeAsS; carente de valor económico), la cual es rechazada en el proceso de concentración por flotación y enviada a la balsa de estériles. Al respecto, cabe mencionar que mientras el arsénico esté fijado a la fase sulfurada, el problema ambiental es mínimo. Por ejemplo, cuando se produjo la rotura del dique contención de la balsa de estériles de Aznalcóllar (Sevilla, España) se realizaron análisis químicos del vertido, detectándose altos valores de arsénico en las zonas contaminadas.
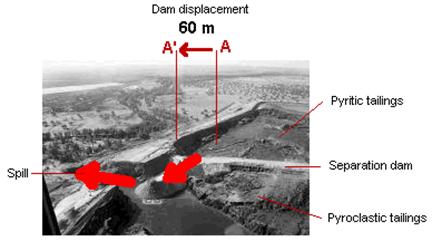
Rotura del dique de contención (flecha roja, izquierda) y derrame de los materiales de la balsa de Aznalcóllar (1998).
Lo que no se dijo, o al menos no se explicitó en los medios de comunicación, es que el vertido no incluía arsénico soluble (como anión complejo), sino que el metal estaba principalmente fijado a la fase sulfurada (arsenopirita). En estos casos la química ambiental puede llevar a fuertes errores conceptuales si carece del obligatorio respaldo mineralógico. Aun si el estudio químico de un vertido se realiza con la mejor intención del mundo, la ignorancia (en el mejor de los casos) resulta inexcusable. ¿Significa esto que el vertido de Aznalcóllar era inocuo? de ninguna manera, como hemos visto en las reacciones anteriores, la arsenopirita acaba oxidándose, y liberando complejos de arsénico:
2 FeAsS + Fe2(SO4)3 → 2 H3AsO4 + 4 FeSO4 + H2SO4
Pero éste no es ni remotamente un proceso rápido, por lo cual la alarma inicial (con respecto a este elemento), inducida en parte por los medios de comunicación y grupos ambientalistas, era en algunos sentidos injustificada, y bastaba con remover la fracción sólida para alejar un peligro futuro cierto de contaminación por arsénico.
Un último apartado guarda relación con el clima. Si hablamos de la química de los procesos de oxidación, y de acidificación de cursos de agua (drenaje ácido), resulta obvio que la mayor o menor cantidad de lluvias o las temperaturas jugarán un papel importante. Por ejemplo, mientras menor la temperatura, más lenta la cinética de las reacciones, lo que significa que los procesos químicos tardan más en completarse. Así, en climas muy fríos los procesos de oxidación son en general poco eficaces. Por otro lado, un exceso de precipitaciones, como en las zonas tropicales, inducirá a una dilución de las soluciones, y por lo tanto la eficacia de los procesos químicos también se verá mermada. Como resulta obvio, precipitaciones muy escasas tampoco favorecerán los procesos de oxidación y lixiviación de metales. De ahí que los climas de tipo mediterráneo a semi-desérticos sean los más adecuados para favorecer este tipo de procesos: temperaturas medias-altas y precipitaciones no demasiado abundantes.
Aunque no se ha mencionado antes, existe otro factor importante en estos procesos: la litología y los minerales de ganga. Por ejemplo, las rocas (e.g. calizas) o minerales carbonatados (e.g. calcita) reaccionarán con las soluciones ácidas neutralizándolas (reacciones solución-roca, solución-mineral consumidoras de hidrogeniones), despojándolas así en gran parte de su capacidad de lixiviación y transporte de metales.
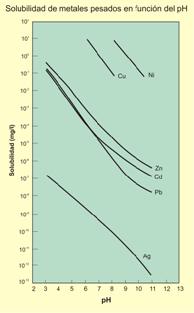
Solubilidad de distintos metales en función del pH del medio acuoso.
El caso del arsénico es particularmente significativo. Se trata de un elemento virtualmente “todoterreno” en los ambientes acuáticos.
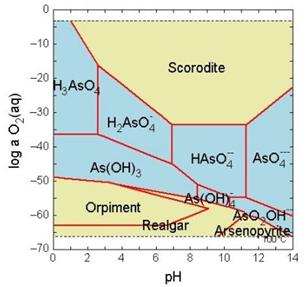
Note el amplio rango de Eh y pH en el cual pueden existir especies solubles de arsénico.
El que el arsénico pueda formar aniones complejos de arsenito (As3+) y arsenato (As5+) le permite desenvolverse en un amplio rango de condiciones de Eh-pH en la naturaleza, siendo su adsorción a partículas coloidales de goethita (u otras especies oxidadas de hierro) la única forma posible de inmovilizar a este elemento.

Goethita del río Toro (Chile) con arsénico adsorbido (zonas claras).
Imagen SEM-EDX.
Hasta aquí se puede crear la impresión (equivocada) de que los principales problemas de contaminación tienen que ver con la minería. Aun cuando la minería constituye la principal fuente de metales de la humanidad (recordemos que una parte se obtiene por reciclado), ni remotamente constituye un tipo de actividad industrial importante, salvo a escala local, o como mucho regional en algunos países. En este sentido recordemos que los problemas de contaminación más serios en los países industrializados se derivan en gran medida de sus industrias (química, farmacéutica, electrónica, etc.), y que dado que éstas muchas veces se localizan cerca del curso de los ríos, causan graves daños al medio ambiente.
Un tipo de actividad que no suele tener “mala prensa” entre la opinión pública, y que no obstante causa graves trastornos al medio ambiente, es la agricultura. Cómo, podría uno preguntarse, una actividad tan apacible (en principio) puede causar tales daños. Existen dos causas directas: los fertilizantes y los pesticidas. La agricultura moderna depende del uso masivo de fertilizantes, siendo los dos más comunes: el super fosfato triple y el nitrato de amonio. El primero es producido a partir de minerales fosfatados tales como el fluorapatito:
3 Ca3(PO4)2·CaF2 + 14 H3PO4 + 10 H2O → 10 Ca(H2PO4)2·H2O + 2 HF
El fósforo es un elemento químico esencial para la vida, ya que interviene en la generación de energía (ciclo de Krebs) a través de la transformación de adenosin trifosfato (ATP) en adenosin difosfato (ADP):
ATP + H2O → ADP + H3PO4 + 34 kJ mol-1
Por otra parte el fósforo es también un constituyente esencial de los ácidos nucléicos (ADN y ARN). El problema ambiental relacionado con los fertilizantes fosfatados radica en su solubilidad, que hace que el fósforo pueda alcanzar los cursos de aguas y lagos. En estos medios acuáticos el principal factor biolimitante es precisamente el fósforo. Si los niveles de este elemento aumentan, quienes sacan más partido son las algas y bacterias, que se reproducen en elevadas cantidades, consumiendo la mayor parte del oxígeno disuelto en las aguas (eutrofización).

Crecimiento desmedido de las algas durante la eutrofización de un lago.
Este proceso conlleva la asfixia y muerte de peces y otros organismos acuáticos. Los problemas ocasionados por el nitrato de amonio (NH4(NO)3) son equivalentes, aunque pueden tener consecuencias aun más graves. Por ejemplo, la inducción de un tipo de anemia en niños pequeños, el denominado síndrome de los niños azules, causado por una reducción en la capacidad de la hemoglobina de transportar oxígeno, debido al exceso de compuestos nitrogenados.
Entre los pesticidas más utilizados se encuentran los denominados organofosfatados (OPs) tales como el paratión y malatión, que han sido utilizados por muchos años como compuestos no persistentes. Aunque están diseñados para romperse en productos solubles no dañinos, algunos OPs son altamente tóxicos y han causado graves daños de salud e incluso la muerte en agricultores.

Aplicación mecanizada de pesticidas en los campos.
2.2.3.- Vertido directo de líquidos
Como comentábamos en otra sección, la adopción de medidas ambientales restrictivas en cuanto a la emisión de SO2 por parte de las fundiciones de sulfuros de metales de base, trajo como consecuencia la producción masiva de ácido sulfúrico en las compañías mineras que empleaban este tipo de procesos metalúrgicos. Aunque la lixiviación in situ de minerales de cobre oxidados por soluciones ácidas era largamente conocida, el exceso de ácido sulfúrico en el mercado potenció la utilización generalizada de este proceso hidrometalúrgico. Los yacimientos de cobre localizados en regiones semi-áridas del mundo suelen presentar una gran variedad de minerales oxidados de cobre en la zona cercana a la superficie. Entre éstos reconocemos sulfatos como la antlerita, carbonatos como la malaquita o la azurita, oxicloruros como la atacamita, y así una extensa lista. Gran parte de estos minerales, especialmente los sulfatos, son fácilmente lixiviables con soluciones aciduladas con ácido sulfúrico. Dicho proceso se realiza sobre una pila de minerales extraídos de la mina, triturados y dispuestos sobre una superficie impermeable inclinada hacia una esquina (heap-leaching). Esta pila de mineral se riega mediante aspersores que distribuyen homogéneamente la solución ácida. El resultado es la puesta en solución de cationes Cu2+, los que junto con la solución pasan a una piscina (tanque; pregnant solution pond) para su extracción por solventes orgánicos.
El proceso se completa por electrorecuperación de la fase metálica (SX-EW).

Una típica instalación de SX-EW (Toquepala, Perú).
Este tipo de operaciones también se realiza en la actualidad sobre minerales sulfurados de cobre, agregándose cepas bacterianas para agilizar el proceso.
Otro proceso minero-metalúrgico equivalente, pero realizado en condiciones de alcalinidad es la cianuración en pila de menas auríferas:
4 Au + 8 CN– + O2 + 2 H2O → 4 [Au(CN)2]– + 4 OH–
Para optimizar las condiciones de operación el pH debe ser mantenido sobre 10.

Cianuración en pila en la mina El Soldado (Chile).
En estas operaciones la solución contiendo el complejo de aurocianuro ([Au(CN)2]–) es pasada por carbono activo (e.g. proceso CIP: carbon in pulp), el que retiene el oro y dejan pasar el cianuro. Alternativamente, en el caso de menas mixtas (con sulfuros y/o sulfosales de plata) resulta más aconsejable utilizar el proceso Merrill-Crowe que utiliza zinc metálico para la precipitación del oro:
2 [Au(CN)2]– + Zn → 2 Au + [Zn(CN)4]2-
El cianuro es utilizado además en otros procesos industriales relacionados con el tratamiento de metales. Este compuesto presenta una alta toxicidad, ya que bloquea encimas vitales relacionadas con el transporte e intercambio de oxígeno a nivel celular. Los principales problemas que pueden ocasionar los derrames involuntarios de soluciones ácidas o cianuradas tienen que ver con un deterioro substancial, aunque transitorio de las aguas, e inducir la muerte de la fauna y flora que albergan. En el caso de las soluciones ácidas, estas acaban por diluirse a niveles de pH aceptables, o bien son neutralizadas por reacciones tipo buffer debido a la presencia de los aniones carbonato y bicarbonato presentes en éstas:
H+ + HCO3– = H2O + CO2
2 H+ + CO32- = H2O + CO2
En el caso del cianuro se produce una degradación natural del compuesto a bicarbonato y amoníaco, ya que inestable en condiciones oxidantes. La reacción la podemos resumir de la siguiente manera:
CN– + 2 H2O + 0.5 O2 → HCO3– + NH3
El amoníaco establece a su vez una reacción de equilibrio en el medio acuoso, formando el ión amonio:
NH3 + H2O = NH4+ + OH–
Aunque la degradación natural del cianuro no es un proceso rápido, éste puede ser acelerado mediante reacciones del tipo:
2 CN– + 5 OCl– + 2 OH– → N2 + 2 CO32- + 5 Cl– + H2O
De cualquier manera, los efectos inmediatos pueden ser dramáticos. Como ejemplo podemos citar el incidente de Baia Mare (Rumania), donde el 30 de Enero del año 2000 se vertieron 130.000 m3 de solución cianurada a los ríos Lupes, Somes, y finalmente al Tisza, y Danubio. El incidente afectó a varios países (ver figura abajo), y entre ellos Hungría pensaba reclamar US$ 100 millones en daños.
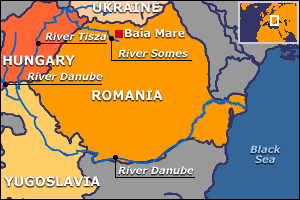
Zona afectada por el incidente de Baia Mare (BBC).
Un tipo de vertido líquido que no podemos dejar de mencionar, aunque sea brevemente, es el de hidrocarburos. Dado el carácter genérico del término deberíamos hacer una distinción en dos grandes categorías: NAPL y DNAPL. Los NAPLs corresponden a la sigla en inglés de Non Aqueous Phase Liquid: fase líquida no acuosa, es decir, líquidos inmiscibles con el agua, y de menor densidad, es decir, suelen ser hidrocarburos derivados del petróleo, que por lo general no tienden a infiltrarse en presencia de agua, debido a que flotan sobre ésta, aunque no obstante pueden introducirse en acuíferos. Los DNAPLs corresponden a la sigla en inglés de Dense Non Aqueous Phase Liquid: fase líquida densa no acuosa, es decir, líquidos inmiscibles con el agua, y de mayor densidad que ésta, que pueden ser de naturaleza diversa, y que constituyen en la actualidad un serio problema por la persistencia y capacidad de infiltración y migración de estos productos en el subsuelo. Algunos de ellos corresponden a disolventes orgánicos, como el tricloroeteno, empleado en tintorería.
El tema de los vertidos de petróleo es quizás uno de los más sensibles en el mundo que rodea a la protección del medio ambiente. Por ejemplo, no hay que olvidar en este sentido la alarma social que causó en España y otros países europeos el vertido derivado del buque tanque Prestige, el que se hundió frente a Finisterre el 19 de Noviembre de 2002, con unos 38.000-50.000 litros de hidrocarburos aun en sus tanques (sin contar el derrame directo que duró días).
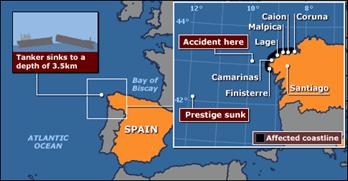
Zona de hundimiento del buque tanque Prestige (el 19.11.02) (BBC).

Efectos de la marea negra.
Nadie puede discutir la importancia vital que tienen los combustibles en el mundo moderno, pero claro está, el trasporte de éstos, desde sus fuentes hasta la gasolinera más cercana del barrio, no está exento de riegos. Los accidentes pueden ocurrir en cualquiera de los siguientes lugares: 1) en las torres de perforación; 2) en los oleoductos (hay más kilómetros de oleoductos que de líneas férreas en el mundo); 3) en las barcazas que transportan petróleo o derivados en los ríos, y en los camiones tanque que surcan prácticamente todas las carreteras del mundo; y 4) de manera notable, en los llamados súper petroleros, que pueden cargar hasta más de 800 “millones” de litros. Aunque estos últimos suelen centrar la atención de los medios de comunicación (y como resultado, de los ciudadanos de un país) en respuesta a una tragedia, se suele olvidar, que las “pequeñas” fugas accidentales y las fugas “crónicas” de combustible causan más daño al medio ambiente (globalmente) que los grandes accidentes. Por ejemplo, la vida útil de una sección de oleoducto es de unos 15 años, sin embargo muchos de éstos sobrepasan esa edad con creces, por lo cual presentan un riesgo cierto de rotura, y por lo tanto de problemas de contaminación. Normalmente los efectos más terribles de un derrame de petróleo se encuentran en el daño directo a la fauna que habita las zonas costeras, y que incluye desde las manchas a los efectos carcinogénicos que pueden presentar algunos componentes del petróleo.

Nutrias mostrando los efectos de la contaminación en sus cuerpos.
Sin embargo, los vertidos de petróleo en zonas continentales también pueden originar daños severos, como el convertir una zona cultivable en un pantano tóxico, o más simplemente, haciendo que el agua deje de ser potable. Esto puede ocurrir tanto a la escala de cursos de superficie, o de las aguas subterráneas, lo cual es aun más grave.
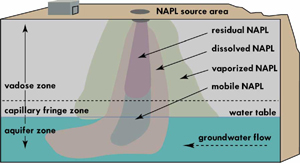
Introducción de NAPLs en un acuífero.
2.3.- Transporte e inmovilización de metales y fases minerales en el medio acuático
Hasta ahora hemos revisado de una manera simple como se incorporan los metales al medio acuático. Dado que los metales poseen la capacidad de movilizarse dentro de éste, la pregunta es “cómo lo hacen”. En nuestro capítulo introductorio a la química de soluciones, analizamos el caso del hierro bajo el punto de vista de las condiciones de Eh-pH del medio, su capacidad para migrar como Fe2+ y su precipitación como especies oxidadas de Fe3+. Por ejemplo, en condiciones de acidez y [Fe2+] = 1M tenemos:
3 H2O + Fe2+ = Fe(OH)3 + 3 H+ E0 = 0.98 v
Eh = 0.98 – 0.177 pH
y para [Fe2+] = 10-7M:
Eh = 1.39 – 0.177 pH
En condiciones básicas tendremos:
Fe(OH)2 + OH– = Fe(OH)3 + e– E0 = -0.56 v
Eh = 0.27 – 0.059 pH
De aquí podríamos deducir directamente que a partir del momento que superamos de manera ascendente la curva que separa [Fe2+] de [Fe3+] (condiciones progresivamente más oxidantes) el hierro precipitará y ya está, fin de la historia. Bajo el punto de vista de la química de soluciones esto es estrictamente correcto, sin embargo, la experiencia de campo nos muestra que el hierro puede efectivamente migrar en condiciones oxidantes. ¿Cómo puede darse tal fenómeno? Para ello tenemos que remitirnos a la físico-química de los sistemas coloidales, y definir primero “qué es una suspensión coloidal y qué es un coloide”. A diferencia de las soluciones “verdaderas”, con iones o iones complejos disueltos en un medio acuoso, un sistema coloidal consiste en partículas muy pequeñas de una fase “dispersas” de forma metaestable en otra; en el caso que nos atañe, dispersas en una fase líquida. El tamaño de partículas en los sistemas coloidales está en el rango de 1-100 nm. Las partículas permanecen en suspensión porque tienen la capacidad de adsorber iones del medio. Por ejemplo, las partículas de SiO2 se recubren de aniones OH–, los que le confieren una carga neta negativa. Dado que todas las partículas de SiO2 presentan la misma carga, estas se repelen entre ellas, manteniéndose así en suspensión.
Volviendo a la pregunta que nos hacíamos antes sobre la capacidad de movilización del hierro oxidado en condiciones de Eh-pH “imposibles”, podemos ahora ofrecer una respuesta plausible. El hierro obviamente no puede migrar como cationes Fe3+, pero sí puede hacerlo como finas partículas de goethita (Fe(3+)OOH) cargadas positivamente. La razón para esta carga radica en el ambiente en que se forman las especies oxidadas de hierro, ricas en hidrogeniones, esto es, en cargas positivas.
La importancia de las dispersiones coloidales es doble, por un lado permiten la migración de cationes o aniones que bajo determinadas condiciones de Eh-pH no sería posible, por otra parte, dada la carga eléctrica de las partículas coloidales, éstas pueden captar (adsorber) aniones o cationes dependiendo del signo de dicha carga. El interés ambiental de este último tipo de procesos queda magníficamente ejemplificado en el caso de las especies solubles de arsénico. Como vimos en otras secciones, el arsénico puede migrar como aniones complejos de arsenito y arsenato, en un amplísimo rango de condiciones de Eh-pH. Sin embargo, en presencia de especies oxidadas de hierro, como la goethita, estas especies de arsénico son adsorbidas, y la razón es simple: las especies aniónicas tienen cargas negativas, mientras que las partículas coloidales de goethita presentan en superficie cargas positivas. Cuando las cargas de una partícula coloidal son neutralizadas ya no puede continuar en la dispersión. En este caso las partículas “floculan”, esto es, se van al fondo dando como producto final un “gel”.
¿De qué dependen las capacidades sorcitivas de las partículas coloidales? La adsorción consiste en la acumulación de una especie del líquido (adsorbato) sobre la superficie de una fase sólida (adsorbente). Es un proceso complejo, en el que se establecen fuerzas de asociación entre ambos componentes de muy diverso tipo. La precipitación superficial (adsorción) consiste en la formación de un precipitado cristalino sobre el sólido, que puede tener su misma u otra composición. El desarrollo de estos fenómenos en su conjunto depende de las características del sorbato y del sorbente en varios aspectos: naturaleza de la fase líquida, de la especie o especies implicadas, de las condiciones físico-químicas del entorno. En concreto, las características de la fase sólida que influyen son el área superficial y la carga eléctrica. El área superficial de una fase mineral a su vez depende de dos factores principales: su tamaño de grano y su naturaleza intrínseca. En lo que se refiere al tamaño de grano, cuanto menor sea, mayor será la superficie específica para un mismo volumen de la fase. En cuanto a la naturaleza de la fase, cada mineral presenta sus propias características en función de caracteres cristalográficos y físico-químicos.
Un grupo de minerales muy importante bajo el punto de vista ambiental, que forma suspensiones coloidales son las arcillas. Las razones para el comportamiento coloidal de éstas radican en su reducido tamaño y su descompensación eléctrica. La descompensación es producida por un exceso de cargas negativas, aunque la carga final dependerá en gran medida del pH del medio, por ejemplo, en condiciones de acidez se puede producir un exceso de cargas positivas.
La adsorción de plomo (metal pesado altamente tóxico) por parte de la goethita puede ser descrita como un proceso del tipo:
Fe-OH + Pb2+ → Fe-OPb+ + H+
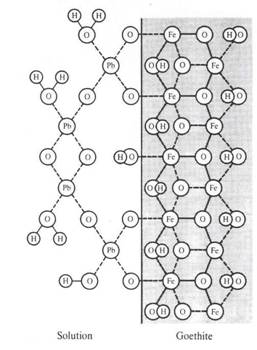
Captación de plomo por la goethita.
La reacción de arriba tiene importancia en lo que se refiere a la fijación del plomo lixiviado de labores mineras o escombreras, un proceso del tipo:
PbS + H2CO3 +2O2 → Pb2+ + 2HCO3– + SO4-2 + 2H+
El plomo así lixiviado puede migrar a pH ácido, sin embargo, a medida que este sube, el plomo va quedando fijado como catión adsorbido en goethita, y cuando se supera un pH de 6, todo el plomo ha sido retenido.
Otro factor que controla la precipitación de los compuestos químicos es el producto de solubilidad (Ksp). Esto es particularmente notable en el caso de las sales (e.g. sulfatos, carbonatos). Como vimos en el capítulo introductorio de química general, cuando se supera el Ksp comienza la precipitación de una sal. Esto puede ocurrir por dos razones: 1) aporte externo de unos de los constituyentes de la sal; y 2) por evaporación progresiva del medio acuoso, que lleva a una subida de la concentración de las especies iónicas disueltas, y como resultado, a una superación del valor de Ksp. Esto último es particularmente significativo en regiones áridas, y aplicable a dos casos concretos, la precipitación de sales de cobre y hierro en balsas de lodo abandonadas, y en la formación de los salares. En el primer caso la evaporación supera a las precipitaciones, lo que conlleva procesos de formación de sales (e.g. sulfatos complejos) en los niveles superiores de una escombrera. Por el contrario, en un clima húmedo las precipitaciones superan a la evaporación, lo que conlleva una lixiviación hacia abajo de cationes tales como Zn2+ o Cu2+.
El caso de la izquierda (clima árido) ilustra el movimiento de soluciones por capilaridad hacia los
niveles superiores de una balsa o escombrera, donde las especies en solución (sulfatos, carbonatos, cloruros)
precipitarán. El caso de la derecha muestra la situación en un clima húmedo, con las soluciones arrastrando
cationes y aniones hacia abajo.
Minerales típicos generados en condiciones áridas son los cloruros, sulfatos y carbonatos básicos (con grupos OH) de cobre tales como la atacamita (Cu2Cl(OH)3), antlerita (Cu3SO4(OH)4) o malaquita (Cu2(CO3)(OH)2) entre muchos otros. La formación de este tipo de minerales es compleja y depende en gran medida de las concentraciones de los siguientes componentes: Cu2+, H+, Cl–, SO42-, y CO2. Lo normal es observar en un mismo afloramiento varios minerales oxidados de cobre, en distintas proporciones. Esto es debido a que es fácil que se generen microambientes químicos, donde ligeras variaciones en los componentes anteriores pueden llevar a la precipitación de un mineral, mientras que al lado, está precipitando otro. Para hacer más compleja la situación, digamos que se también se pueden producir reacciones que llevan a la desaparición de una fase y la aparición de otra, en función de las concentraciones de los componentes del sistema. Por ejemplo, un aumento en CO2 en las aguas hará que la malaquita no sea estable y se forme azurita:
3 Cu2CO3(OH)2 + CO2 → 2 Cu3(CO3)2(OH)2 + H2O
Así mismo, la presencia de iones Cl– pueden llevar a la desaparición de brochantita y la formación de para-atacamita:
Cu4SO4(OH)6 + 2 Cl– → Cu4Cl2(OH)6 + SO42-
El otro caso que hemos mencionado es el de los salares, ejemplificados por los del tipo Andino, que pueden ser encontrados en Chile (e.g. Salar de Atacama) o Bolivia (e.g. Salar de Uyuni). Los salares están constituidos principalmente por carbonatos, sulfatos y cloruros, normalmente dispuestos en ese orden desde afuera hacia el núcleo. La razón de esta tonalidad concéntrica es simple y guarda razón con el producto de solubilidad de estas especies. Analicemos el caso del carbonato y sulfato de calcio (en equilibrio):
[Ca2+]x[CO32-] = 5 x 10-9
[Ca2+]x[SO42-] = 6 x 10-5
Como podemos apreciar, el Ksp del carbonato de calcio es mucho más bajo que el del sulfato. Esto significa que si se produce el más ligero incremento en la concentración de calcio en el sistema, la primera especie en precipitar será el carbonato. Así, en una cuenca con aporte de calcio, los primeros precipitados químicos corresponderán a la fase carbonatada. Si el sistema sigue en evaporación, y no todo el calcio ha sido consumido (o si el aporte de calcio es continuo), la segunda especie en precipitar será el sulfato de calcio. De esta manera empezamos a configurar un patrón concéntrico. El cloruro de sodio tiene un Ksp altísimo, en el orden de 36 (compare con los del sulfato y carbonato de calcio), de tal manera que la evaporación tiene que ser prácticamente total para que empiece a precipitar el cloruro de sodio, lo cual ocurrirá en las etapas póstumas de la cuenca, en su sector central.

Precipitación activa de sales en el Salar de Atacama (Antofagasta, Chile).
La situación en los sistemas salinos es obviamente más compleja, ya que se trata de sistemas químicos de muchos constituyentes, en los cuales la solubilidad es dependiente de las distintas concentraciones. Por ejemplo, la concentración de cloruro de sodio controla la solubilidad del sulfato de calcio. Altas concentraciones de cloruro de sodio disminuyen la solubilidad del sulfato de calcio, mientras que lo inverso ocurre a bajas concentraciones.
Solubilidad del sulfato de calcio en función de la concentración de cloruro de sodio.
Observe las curvas roja y verde (25º y 32ºC, ambientes de superficie).
Para bajas concentraciones de NaCl la solubilidad del sulfato de calcio se incrementa
(pendiente positiva). Esta tendencia se invierte a partir de una concentración de
10 % de NaCl. Note como esta tendencia se modifica además a medida que
aumenta la temperatura en el sistema.
2.4.- Suelos: la vulnerable piel protectora del planeta
El suelo es por principio uno de los sitios principales donde van a parar gran parte de los desechos sólidos y líquidos de cualquiera actividad humana.

Vertido incontrolado de residuos urbanos.
Indiquemos no obstante, que los suelos son también el receptáculo natural de los deshechos no deseables de origen geológico, por ejemplo, de las aguas ácidas con metales pesados provenientes de mineralizaciones sulfuradas aflorantes. Todo lo que no es de utilidad en los procesos industriales, mineros, urbanos, agrícolas, etc., se acumula en el suelo, en muchos casos sin mayores precauciones. Con ello, la escombreras mineras, los residuos de una fábrica, incluyendo líquidos, se han venido depositando sobre los suelos sin control alguno a lo largo de siglos e incluso milenios (recordemos los desechos urbanos y mineros de la Roma clásica). Por otra parte, la actividad agrícola se ha venido enfrentando durante las últimas décadas a la necesidad de aumentar la producción. Esto se ha conseguido sobre dos bases principales: el abonado con productos químicos (nitratos, fosfatos), y el control de plagas. En definitiva, las actividades industriales y mineras por un lado, y las agrícolas por otro, han dado origen al problema de que muchos productos de origen humano, o formados gracias a las actividades humanas, han ido a parar a los suelos, generando a su vez otros problemas: la contaminación de aguas subterráneas, una menor productividad agrícola, la contaminación de cultivos, y el envenenamiento de ganado, afectando de forma directa, y en mayor o menor grado en casa caso, a la economía y a la salud humanas.
Como hemos podido ver en las secciones anteriores, la industria en su conjunto produce toda una serie de contaminantes gaseosos, líquidos y sólidos, que en muchas ocasiones van a parar directamente a los suelos. Esto sucede ya sea por depósito a partir de la atmósfera como partículas sedimentadas o traídas por las aguas de lluvia, por el vertido directo de los productos líquidos de la actividad minera y metalúrgica, o por la infiltración de productos de desechos industriales. Por ejemplo, en el caso de la minería tenemos las aguas provenientes de minas a cielo abierto, escombreras (mineral dumps), o talleres de la mina u otras edificaciones más o menos contaminantes en cada caso.

Contaminación de suelos con drenaje ácido de mina.
Uno de los principales problemas que puede producir la minería es la adición al suelo de una fase líquida. Esta habitualmente presenta una composición muy diferente a la que habitualmente se infiltra en el mismo en ausencia de actividades mineras (agua de lluvia). Las interacciones resultantes pueden ser muy variadas en función de la composición química del fluido, la mineralogía del suelo, y el factor climático (temperaturas medias, abundancia y frecuencia de lluvias). Los efectos en el suelo en relación con la presencia de contaminantes pueden también ser variados. En unos casos los contaminantes se acumulan en formas de alta solubilidad, de forma que están disponibles para que los animales y vegetales que viven sobre el mismo los ingieran. De la misma manera se puede producir la contaminación de los acuíferos, ya que las aguas de infiltración pueden incorporar a éstos los contaminantes. Los suelos también pueden tener un efecto absorbente, actuando como un biofiltro altamente reactivo que facilita la inmovilización de los contaminantes gracias a procesos físicos (filtración), físico-químicos (neutralización, adsorción, absorción, precipitación, complejación) o biológicos (biodegradación). En este sistema juegan un papel especialmente importante las arcillas, debido a sus propiedades de adsorción. Recordemos que la capacidad de captación iónica varía de una arcilla a otra y se relaciona directamente con las características estructurales de éstas. En el tipo 1:1 el armazón TO-TO se sustenta mediante enlaces tipo puente de hidrógeno, entre los grupos (OH) de las capas octaédricas y los oxígenos de las tetraédricas. Este enlace es lo suficientemente fuerte como para mantener la estructura global y al mismo tiempo impedir la entrada de cationes entre capas. Por el contrario, en las arcillas 2:1 (TOT-TOT) este tipo de enlace entre capas no es posible (se enfrentan capas tetraédricas). En este caso el armazón estructural se compensa eléctricamente introduciendo cationes interlaminares. En las esmectitas se producen múltiples substituciones en las capas octaédricas y tetraédricas, que le confieren una carga global negativa. Sin embargo, esta carga es relativamente débil, razón por la cual no se pueden ligar cationes de una manera fuerte en el espacio interlaminar. De esta manera, se permite la entrada de agua y otras moléculas que causan un aumento en el espacio interlaminar (“hinchamiento” de la arcilla). Con el agua vienen cationes disueltos, que su vez, pueden ser fijados en dicho espacio. De esta manera las arcillas esmectíticas actúan como depuradoras naturales de los suelos.
Estructura de las arcillas tipo esmectita. Note el espaciado interlaminar donde se pueden acomodar cationes.
Otra labor importante de depuración de soluciones viene dada por las partículas minerales con comportamiento coloidal, como la goethita (carga positiva), con capacidad para fijar complejos aniónicos arseniacales. Otros óxidos, como los de manganeso constituyen coloides de carga negativa, que les permite fijar cationes en solución.

Suelo rojo, caracterizado por la presencia de fases oxidadas de hierro.
Sin embargo, cuando se supera la capacidad de amortiguación geoquímica del suelo, éste se convierte de hecho en fuente de contaminación. De igual forma, un cambio en las condiciones climáticas puede producir la reversibilidad del proceso. Por ello a menudo se habla de que la presencia de contaminantes en el suelo constituye una bomba de tiempo química, que aún si en un determinado momento no produce efecto alguno, si puede hacerlo en un futuro.
Recordemos que la erosión del suelo conlleva un transporte de los contaminantes a otras áreas. Esto nos lleva a otros conceptos importantes en lo relativo a la presencia de contaminantes en el suelo: los de geodisponibilidad y biodisponibilidad. La geodisponibilidad es la consecuencia directa de la actividad minera: al llevar a cabo la explotación minera de un yacimiento, se ponen a disposición del medio geológico unos elementos que antes no lo estaban, o lo estaban de forma mucho más limitada. Cabe destacar, no obstante, que muchos yacimientos minerales, particularmente los de menas sulfuradas, son en sí fuentes naturales de contaminación ambiental. Esto depende en gran medida de si son o no aflorantes, de su profundidad (en especial, si se localizan por encima o por debajo del nivel freático), composición mineralógica, etc. La biodisponibilidad, por su parte, sería “el grado por el cual un contaminante en una fuente potencial, está disponible para ser tomado por un organismo”. Por ejemplo, muchas plantas tienen la capacidad de absorber determinadas concentraciones de elementos pesados, siempre que se encuentren en el suelo en formas solubles, o asociados a nutrientes básicos.
2.5.- Bibliografía
Andrews, J.E., Brimblecombe, P., Jickells, T.D., y Liss, P.S. 1996. An introduction to environmental chemistry. Blackwell Science, Oxford, 209 pp.
BBC, 2001. One year on: Romania’s cyanide spill. http://news.bbc.co.uk/2/hi/europe/ 1146979.stm
BBC, 2002. Major new slick hits Spain coast. http://news.bbc.co.uk/2/hi/europe/ 2532013.stm
Chakravarty, S., Dureja, V., Bhattacharyya, G., Maity, S., y Bhattaacharjee, S. 2002. Removal of arsenic from groundwater using a low cost ferruginous manganese ore. Water Research, 36: 625-632.
Elliot, D. 1999. Primary brine treatment operations. 1999 Eltech Chlorine/Chlorate Seminar, “Technology Bridge to the New Millenium”, Cleveland, Ohio, 21 pp.
Epstein, P.R., y Selber, J. 2004. Oil a life cycle analysis of its health and environmental impacts (Executive Summary, PDF). http://www.med.harvard.edu/chge/oil.html
Epstein, P.R., y Selber, J. 2004. Oil a life cycle analysis of its health and environmental impacts (Full Report, PDF). http://www.med.harvard.edu/chge/oil.html
Espi, J.A. (Ed.) 2001. El libro de la minería del oro en Iberoamérica. CYTED, Madrid, 398 pp.
Gill, R. 1996. Chemical fundamentals of geology. Chapman & Hall, London, 290 pp.
Higueras, P., Oyarzun, R., Biester, H., Lillo, J., y Lorenzo, S. 2003. A first insight into mercury distribution and speciation in soils from the Almadén mining district. Journal of Geochemical Exploration, 80: 95-104.
Jambor, J.L., Blowes, D.W., y Ritchie, A.I.M. (Eds.) 2003. Environmental aspects of mine wastes. Mineralogical Association of Canada, Vancouver, 430 pp.
Krauskopf, K.B. 1979. Introduction to geochemistry. McGraw-Hill, NY, 617 pp.
Krauskopf, K.B., y Bird, D.K. 1995. Introduction to geochemistry. McGraw-Hill, NY, 647 pp.
NASA 2004. Visible Earth. http://visibleearth.nasa.gov/
López García, J.A. 2004. Gossans. http://www.ucm.es/info/crismine/gossan/ gossanapuntes2.htm
López García, J.A. 2004. Salida de campo del Grupo B de Recursos Minerales a La Unión-Sierra de Cartagena: mineralizaciones de Pb-Zn-Ag, Fe, Sn. http://www. ucm.es/info/crismine/Fotos_Pepe_2/Cartagena_LaUnion.htm.
McLean, J.E., y Bledsoe, B.E. 1992. Behaviour of metals is soils. USEPA Ground Water Issue, EPA/540/S-92/018.
Petrucci, R.H., y Harwood, W.S. 1993. General chemistry: principles and modern applications. MacMillan, NY, 1000 pp.
Porta, J., López-Acevedo, M., y Roquero, C. 1999. Edafología para la agricultura y el medio ambiente. Ediciones Mundi-Prensa. 849 pp.
Smedley, P.L., y Kinniburgh, D.G. 2002. A review of the source, behavior and distribution of arsenic in natural waters. Applied Geochemistry, 17: 517-568.
Smith, J.V. (Ed.) 1999. Colloquium on geology, mineralogy, an human welfare. National Academy of Sciences, Washington DC, 96: 3348-3485.
Soil Survey Division, Natural Resources Conservation Service, United States Department of Agriculture. Official Soil Series Descriptions [Online WWW]. Available URL: «http://www.statlab.iastate.edu/soils/osd/» [Accessed 23 Mar 2001].
Stip, S.L.S.; Hansen, M., Kristensen., Hochella, M.F., Bennedsen, L., Dideriksen, K., Balic-Zunic, T., Léonard, D., y Mathieu, H.J. 2002. Behavior of Fe-oxides relevant to contaminant uptake in the environment. Chemical Geology, 190: 321-337.
USGS, 2004. Glacial Ice Cores Reveal A Record of Natural and Anthropogenic Atmospheric Mercury Deposition for the Last 270 Years. USGS Fact Sheet FS-051-02. http://toxics.usgs.gov/pubs/FS-051-02/
Woods, T.L., y Garrels, R.M. 1986. Phase relations of some cupric hydroxy minerals. Economic Geology, 81: 1989-2007.
Williams, I. 2001. Environmental chemistry. John Wiley & Sons, NY, 388 pp.
3.- Mineralogía y procesos de contaminación de suelos

Típico suelo rojo tropical, con fragmentos de roca.
Los suelos constituyen la superficie del planeta, y como tales, soportan la mayor parte de las actividades humanas, construcciones, infraestructuras, excavaciones mineras, acumulación de residuos procedentes de todo tipo de actividades, etc. Tradicionalmente se consideró que el suelo “lo aguantaba todo”, que se podía verter sobre él todo lo que se desease, que tenía una capacidad de absorción y purificación prácticamente infinita.

Un mundo industrial sin control…
Esto no era más que una verdad a medias. Las capacidades depuradoras de los suelos existen, pero actúan tan a largo plazo que es necesario considerar que a la escala de tiempo humano la regeneración de los suelos no se produce a un ritmo suficiente como para impedir graves problemas de contaminación. En este sentido son necesarias políticas preventivas, para evitar que esta contaminación se produzca, y medidas correctoras, que permitan recuperar lo más rápidamente posible los suelos afectados por la contaminación. La necesidad de protección del suelo se puso de manifiesto en 1972 por el Consejo de Europa en su Carta Europea del Suelo, donde se establecieron los principios generales de protección de éstos, pasando a ser considerados como un recurso no renovable.
En el presente tema estudiaremos el suelo desde el punto de vista de su origen y características, y analizaremos los procesos implicado en la contaminación del mismo.
Estructura del Tema:
3.1.- Origen del suelo
3.2.- Formación de arcillas en el suelo
3.3.- Mineralogía y físico-química del suelo
3.3.1.- Constituyentes de origen mineral
3.3.2.- El agua en los suelos
3.3.3.- Gases en el suelo
3.3.4.- Materia orgánica
3.4.- Contaminantes en el suelo
3.5.- Vulnerabilidad del suelo ante los contaminantes químicos
3.6.- Bibliografía
3.2.- Formación de arcillas en el suelo
Una cuestión importante en la formación del suelo es la génesis de los minerales más característicos del mismo: las arcillas. En concreto, la formación de los minerales de la arcilla en este ambiente está íntimamente ligada a reacciones de hidrólisis de los minerales silicatados de las rocas. Estas reacciones pueden desarrollarse en el medio hidrotermal (durante la formación de un depósito mineral), o como procesos exógenos (bajo condiciones atmosféricas), una vez que las rocas por erosión se encuentran en la superficie o su proximidad. El CO2 disuelto en el agua de lluvia o de los ríos puede desencadenar una serie de procesos hidrolíticos:
CO2 + H2O ® H2CO3
El ácido carbónico así formado reacciona con los feldespatos, induciendo la formación de minerales del grupo de la arcilla. A continuación ilustramos este tipo de reacciones con tres ejemplos conducentes a la formación de caolinita, Al2Si2O5(OH)4:
1) Hidrólisis de anortita (plagioclasa cálcica):
CaAl2Si2O8 + 2 H2CO3 + H2O ® Ca2+ + 2 HCO3– + Al2Si2O5(OH)4
2) Hidrólisis de la albita (plagioclasa sódica):
2 NaAlSi3O8 + 2 H2CO3 + 9 H2O ® 2 Na+ + 2 HCO3– + Al2Si2O5(OH)4 + 4 H2SiO4
3) Hidrólisis de la ortoclasa (feldespato potásico):
2 KAlSi3O8 + 2 H2CO3 + 9 H2O ® 2 K+ + 2 HCO3– + Al2Si2O5(OH)4 + 4 H2SiO4

Roca alterada a arcillas caoliníticas. Note el blanqueo de la roca.
El clima, a través de los parámetros de humedad y temperatura, controla fuertemente el proceso formador de arcillas a partir de los silicatos. Así, en condiciones de humedad y calor la hidrólisis dará lugar a arcillas caoliníticas, e incluso a un residuo final de gibbsita, Al(OH)3. Por el contrario, en climas áridos la arcilla predominante resulta ser del tipo illita-esmectita. Cabe destacar, no obstante, que el mundo de los procesos formadores de arcillas es extraordinariamente complejo, por lo cual lo anteriormente dicho tiene que ser tomado únicamente en el contexto de una simplificación didáctica. Las figuras adjuntas ilustran la complejidad de variables implicadas.
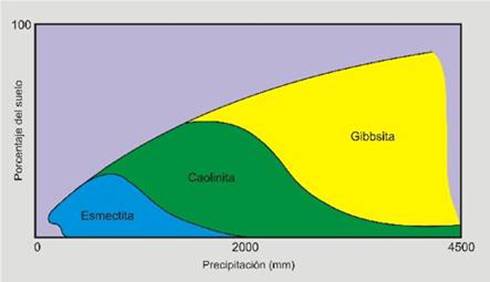
Mineralogía de arcillas formada en el suelo en función de las precipitaciones.
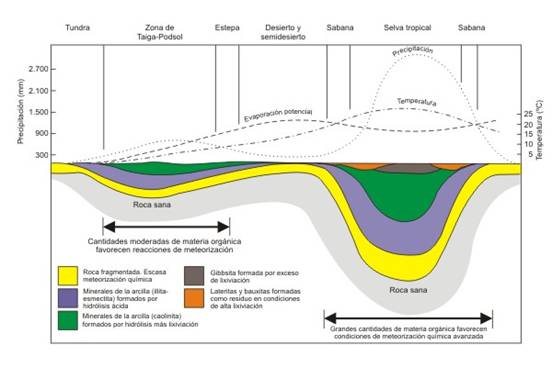
Tipos y espesores característicos de suelos formados en las distintas zonas climáticas.
3.3.- Mineralogía y físico-química del suelo
Como consecuencia del proceso de edafogénesis tenemos un suelo estructurado, en el que cada capa u horizonte presenta unas peculiaridades composicionales, tanto en lo que se refiere a sus componentes mineralógicos como en su textura, y físico-química. Cuatro son, en concreto, los principales componentes del suelo:
1.- Minerales
2.- Agua
3.- Gases
4.- Materia orgánica
3.3.1.- Constituyentes de origen mineral
Los minerales que componen el suelo pueden ser tan variados como lo sea la naturaleza de las rocas sobre las que se implanta. No obstante, hay una tendencia general de la mineralogía del suelo hacia la formación de fases minerales que sean estables en las condiciones termodinámicas del mismo, lo cual está condicionado por un lado por el factor composicional, y por otro por el climático, que condiciona la temperatura, la pluviosidad, y la composición de las fases líquida y gaseosa en contacto con el suelo.
Los minerales del suelo pueden ser de dos tipos: 1) heredados, es decir, procedentes de la roca-sustrato que se altera para dar el suelo, que serán minerales estables en condiciones atmosféricas, resistentes a la alteración físico-química; y 2) formados durante el proceso edafológico por alteración de los minerales de la roca-sustrato que no sean estables en estas condiciones. Los más importantes, y los condicionantes para su presencia en el suelo serían los siguientes:
ü Cuarzo. Es un mineral muy común en los suelos, debido a: 1) su abundancia natural en la mayor parte de las rocas; y 2) su resistencia al ataque químico. El cuarzo confiere al suelo buena parte de su porosidad, debido a que suele estar en forma de granos más o menos gruesos, lo que permite el desarrollo de la porosidad intergranular. Además, es un componente muy inerte, muy poco reactivo, del suelo. Suele encontrase en suelos poco estructurados de textura arenosa.

Típico aspecto de un suelo arenoso.
ü Feldespatos. Suelen ser componentes minoritarios, heredados o residuales de la roca sobre la que se forma el suelo, pues son metaestables en medio atmosférico, tendiendo a transformarse en minerales de la arcilla. Al igual que el cuarzo, conforman la fracción arenosa del suelo, si bien en este caso le confieren una cierta reactividad.
ü Fragmentos de roca. Junto con los dos componentes anteriores, conforman la fracción comúnmente más gruesa del suelo, si bien es este caso el tamaño de fragmentos suele ser superior a 2 cm, de forma que el cuarzo y feldespatos suelen constituir la fracción arenosa del suelo, mientras los fragmentos de roca constituyen la fracción de tamaño grava. La naturaleza de los fragmentos está directamente relacionada con la de la roca sobre la que se forma, si bien ocasionalmente el suelo puede contener fragmentos de origen “externo”, como consecuencia de procesos de transporte y depósito contemporáneos con la formación del suelo. En cualquier caso, son siempre heredados, y nos permiten identificar si el proceso de edafogénesis ha tenido o no aportes externos.
Típico suelo rojo tropical, con fragmentos de roca.
ü Minerales de la arcilla. Son minerales también muy abundantes en el suelo, constituyendo la matriz general del mismo, la componente intergranular entre la fracción arenosa y los fragmentos de roca. Son minerales que proceden de la alteración de los que componen la roca sobre la que se producen los procesos de meteorización, y en función de ello pueden ser muy variados: 1) la illita (equivalente arcilloso de la mica blanca, moscovita), que se forma a partir de feldespatos y micas de rocas ígneas, sedimentarias o metamórficas; 2) la clorita, que se forma a partir de los minerales ferromagnesianos que pueda contener la roca: biotita, anfíbol, piroxeno, olivino; 3) la pirofilita, que puede formarse a partir de minerales ricos en aluminio en la roca original; 4) menos comunes son los filosilicatos del grupo de las arcillas especiales (esmectita-bentonita, sepiolita, palygorskita), que se forman bajo condiciones climáticas muy específicas, o a partir de rocas de composición muy determinada, y que por sus características especiales confieren al suelo propiedades mecánicas diferentes a las habituales (suelos expansivos, suelos instables). Los minerales de este grupo juegan un papel muy importante en la textura y en la físico-química del suelo, pues le confieren plasticidad, impermeabilidad, así como otras propiedades mecánicas y de relación entre el suelo y el agua que contiene, en especial en cuanto a la capacidad de sorción e intercambio iónico que pueda presentar. Hablamos en mayor detalle de estas capacidades más adelante.

Suelo arcilloso.
ü Carbonatos. Los carbonatos son minerales frecuentemente formados por el proceso de edafogénesis, aunque debido a su alta solubilidad su acumulación no suele producirse en el horizonte más superficial. De hecho, los carbonatos pueden formarse en los horizontes A o C, pero su acumulación efectiva se produce solo en el horizonte B o de acumulación, como consecuencia de los procesos de intercambio que se producen en el mismo. Una excepción corresponde a los suelos de regiones de climatología semiárida y con abundantes rocas carbonatadas. En estas regiones, los procesos de intercambio con el suelo suelen ser “en ascenso”: las aguas subterráneas ricas en carbonatos ascienden hasta la superficie del terreno por capilaridad o por gradiente de humedad, depositando ahí los carbonatos, y originando los denominados “caliches”, auténticos escudos de color blanco que recubren la superficie del suelo, como por ejemplo ocurre en buena parte de La Mancha.

Suelos con caliche, note el color blanco del mismo.
ü Óxidos e hidróxidos de hierro, manganeso y aluminio. Los óxidos e hidróxidos de Fe3+ (y a menudo los de aluminio y los de manganeso) son minerales que se suelen acumular en el suelo como consecuencia de procesos de alteración de otros minerales, constituyendo la fase estable del hierro en superficie o condiciones cercanas a la superficie. Se acumulan en forma de agregados: 1) limonita (agregado de óxidos e hidróxidos de Fe), 2) bauxita (de óxidos e hidróxidos de aluminio); y 3) wad (óxidos e hidróxidos de manganeso). Desde el punto de vista estrictamente químico son muy estables, poco o nada reactivos, pero presentan propiedades sorcitivas que hacen que su presencia en el suelo tenga implicaciones físico-químicas notables. Los suelos ricos en óxidos e hidróxidos de hierro, formados por un lavado casi total de otros constituyentes, reciben el nombre de lateritas. Se reconocen por su intenso color rojo y se forman en climas tropicales.

Suelo laterítico en el Complejo de Moa (Cuba).
ü Sulfatos. La presencia de sulfatos en el suelo suele tener la doble vertiente de que pueden ser minerales relativamente comunes, pero al ser compuestos de solubilidad relativamente alta, su acumulación efectiva solo puede producirse bajo condiciones muy determinadas: abundancia de sulfatos (e.g., yesos) en el entorno inmediato, y clima árido o semiárido. En estas condiciones, y al igual que los carbonatos, los sulfatos podrán acumularse en el horizonte B, o en el A, en este segundo caso en forma de costras o eflorescencias (rosas del desierto).
ü Otros minerales. Aparte de los descritos, el suelo puede contener una amplia gama de minerales, en unos casos heredados, en otros formados, todo ello en función de los condicionantes ya mencionados: naturaleza de la roca-sustrato, y factores climáticos. Su importancia e interés pueden ser muy variables.
3.3.2.- El agua en los suelos
Con la excepción de las regiones extremadamente áridas, el agua es siempre un componente del suelo, encontrándose en éstos en forma de humedad intergranular o como hielo (suelos tipo permafrost), en mayor o menor abundancia en función de factores diversos. Debido a la propia dinámica del suelo, el agua siempre contiene componentes diversos en solución, y ocasionalmente también en suspensión, si bien la ausencia de una dinámica de consideración minimiza este último componente.
En función de la naturaleza y textura del suelo el agua puede encontrarse bien como fase libre, móvil en el suelo (en suelos con altas porosidades y permeabilidades), o bien como fase estática (ab/ad sorbida), en los suelos de naturaleza más arcillosa. En el primer caso el agua podrá tener una cierta dinámica, que mantendrá una cierta homogeneidad composicional, mientras que en el segundo caso podrán darse variaciones composicionales más o menos importantes.
El agua en el suelo suele tener una dinámica bidireccional: el agua de lluvia o de escorrentía, por lo general poco cargada en sales (aunque no siempre), se infiltra desde superficie, y puede producir fenómenos de disolución, hidrólisis y/o precipitación de las sales que contiene. Por ejemplo, el CO2 atmosférico induce la formación de ácido carbónico: CO2 + H2O ® H2CO3, que a su vez induce la disolución de carbonatos: CaCO3 + H2CO3 ® Ca2+ + 2HCO3–. En épocas secas se produce el fenómeno inverso, y las aguas contenidas en los acuíferos tienden a subir por capilaridad o por gradiente de humedad hasta la superficie, donde se produce su desecación, de forma que durante este proceso de ascenso tienden a perder por precipitación las sales que contienen en disolución. Este proceso puede tener consecuencias desastrosas cuando interviene la mano del hombre, por ejemplo con irrigación de suelos en zonas áridas-semiáridas, con consecuencias de salinización extrema. Ejemplos dramáticos de estos fenómenos se encuentran en algunas regiones de Australia y se están comenzando a observar en Almería debido a la descontrolada actividad agrícola.

Salinización de suelos.
La composición del agua contenida en el suelo, en cuanto a su contenido en sales solubles (bicarbonatos, carbonatos, sulfatos, cloruros) estará condicionada, como la mineralogía, por factores de la litología del suelo y su entorno, y por factores climáticos. La proximidad de explotaciones mineras de minerales metálicos sulfurados condicionará por lo general un alto contenido en sulfatos, y a menudo en metales pesados.
3.3.3.- Gases en el suelo
El suelo a menudo contiene gases, que pueden tener procedencias diversas: 1) aire atmosférico, que se infiltra desde superficie; 2) gas liberado durante alguna reacción, ya sea estrictamente química: CO2 liberado por la descomposición de carbonatos en medio ácido; o bioquímica: gases metabólicos de microorganismos: CH4, CO2; y 3) ocasionalmente puede contener también radón, gas noble radioactivo que se produce como consecuencia de la fisión natural de isótopos de potasio, torio o uranio, acumulándose sobre todo en los suelos de las áreas graníticas. Estos gases pueden encontrarse en disolución en el agua intersticial, no como fase libre.
3.3.4.- Materia orgánica
La materia orgánica que contiene el suelo procede tanto de la descomposición de los seres vivos que mueren sobre ella, como de la actividad biológica de los organismos vivos que contiene: lombrices, insectos de todo tipo, microorganismos, etc. La descomposición de estos restos y residuos metabólicos da origen a lo que se denomina humus. En la composición del humus se encuentra un complejo de macromoléculas en estado coloidal constituido por proteínas, azúcares, ácidos orgánicos, minerales, etc., en constante estado de degradación y síntesis. El humus, por tanto, abarca un conjunto de sustancias de origen muy diverso, que desarrollan un papel de importancia capital en la fertilidad, conservación y presencia de vida en los suelos. A su vez, la descomposición del humus en mayor o menor grado, produce una serie de productos coloidales que, en unión con los minerales arcillosos, originan los complejos organominerales, cuya aglutinación determina la textura y estructura de un suelo. Estos coloides existentes en el suelo presentan además carga negativa, hecho que les permite absorber cationes H+ y cationes metálicos (Ca2+, Mg2+, K+, Na+) e intercambiarlos en todo momento de forma reversible; debido a este hecho, los coloides también reciben el nombre de complejo absorbente.

Horizonte rico en humus (color negro) en un suelo.
Otro dato relevante con respecto a la materia orgánica es su afinidad por los metales pesados. Cuando éstos se encuentran en disolución, a menudo forman complejos orgánicos solubles, que pueden polimerizarse sobre los complejos moleculares del humus. También pueden formar directamente complejos insolubles con los compuestos del humus. De esta forma, la materia orgánica del suelo a menudo actúa como almacén de estos elementos, si bien puede transferirlos a la vegetación o a la fase acuosa si se produce su descomposición en medio ácido u oxidante.
Otro componente orgánico de los suelos es el ácido fúlvico, que es un tipo de ácido húmico débilmente polimerizado, que interviene en el proceso de podsolización. Junto con las arcillas y el hierro presentes en el suelo, este ácido forma complejos coloidales que por lixiviación son desplazados hasta cierta profundidad, donde finalmente floculan como consecuencia de actividad bacteriana.
Estructura química del ácido fúlvico.
3.4.- Contaminantes en el suelo
El suelo es, por principio, el sitio donde van a parar gran parte de los desechos sólidos y líquidos de cualquier actividad humana. Indiquemos no obstante, que los suelos son también el receptáculo de los deshechos no deseables de origen geológico, por ejemplo, de las aguas ácidas con metales pesados provenientes de mineralizaciones sulfuradas aflorantes.
Todo lo que no es de utilidad en los procesos industriales, mineros, urbanos, agrícolas, etc., se acumula en el suelo, en general sin mayores precauciones. Con ello, la escombreras mineras, los productos producidos en una fábrica, muchos desechos líquidos, se han venido depositando sobre los suelos sin control alguno a lo largo de siglos e incluso milenios (recordemos los desechos urbanos y mineros de la Roma clásica).

Vertido incontrolado de basura.
Por otra parte, la actividad agrícola se ha venido enfrentando durante las últimas décadas a la necesidad de aumentar la producción, sobre dos bases principales: el abonado, y el control de plagas. Baste mencionar dos ejemplos: el uso intensivo de nitratos y fosfatos. En países como Reino Unido y Francia existen serios problemas de contaminación de acuíferos con compuestos nitratados.

Nitrato natural de Chile para fertilizantes.
En definitiva, las actividades industriales y mineras por un lado, y las agrícolas por otro, han dado origen al problema de que muchos productos de origen humano, o formados gracias a las actividades humanas, han ido a parar a los suelos, generando a su vez otros problemas: la contaminación de aguas subterráneas, la bajada de productividad agrícola, la contaminación de cultivos, el envenenamiento de ganado, la contaminación de la selva, afectando así de forma directa la salud humana y ambiental.

Derrame en la selva tropical de un concentrado de sulfuros de cobre, el cual se ha oxidado (OK Tedi, Papua Nueva Guinea).
A pesar del impacto directo de los contaminantes sobre el suelo, el interés social sobre la protección (y aún más, sobre la recuperación) de éste ha sido muy posterior al manifestado por el aire y el agua, probablemente porque los efectos de esta contaminación no son tan evidentes, sino que se ponen de manifiesto a más largo plazo.
En la actualidad, la legislación medioambiental en los países desarrollados pone especial énfasis en la multifuncionalidad del sistema suelo-agua, considerando como sus principales funciones las relativas a: medio y soporte de transporte, filtro de agua, crecimiento vegetal y medio participativo en los ciclos bioquímicos. Las medidas de protección del suelo están orientadas a la prevención de la contaminación local fomentando las medidas de aislamiento y control, así como la reglamentación de emisiones aceptables para contaminación difusa que aseguren el cumplimiento de las funciones del suelo. En los países más avanzados se trabaja en la línea de intentar asegurar la recuperación de los suelos afectados por el problema, impidiendo la venta de terrenos hasta tanto el propietario actual no lleve a cabo esta tarea, de forma que el problema no quede bloqueado por una transferencia de propiedad que diluya responsabilidades.
3.5.- Vulnerabilidad del suelo ante los contaminantes químicos
Los suelos poseen un efecto absorbente, actuando como un filtro altamente reactivo que facilita la inmovilización de los contaminantes gracias a procesos físicos (filtración), físico-químicos (neutralización, adsorción, absorción, precipitación, complejación) o biológicos (biodegradación). Pero claro está, esto significa que el suelo “acumula” muchos de los contaminantes. En este sistema juegan un papel especialmente importante las arcillas, debido a sus propiedades de adsorción. Recordemos que la capacidad de captación iónica varía de una arcilla a otra y se relaciona directamente con las características estructurales de éstas. En el tipo 1:1 el armazón TO-TO se sustenta mediante enlaces tipo puente de hidrógeno, entre los grupos (OH) de las capas octaédricas y los oxígenos de las tetraédricas. Este enlace es lo suficientemente fuerte como para mantener la estructura global y al mismo tiempo impedir la entrada de cationes entre capas. Por el contrario, en las arcillas 2:1 (TOT-TOT) este tipo de enlace entre capas no es posible (se enfrentan capas tetraédricas). En este caso el armazón estructural se compensa eléctricamente introduciendo cationes interlaminares. En las esmectitas se producen múltiples substituciones en las capas octaédricas y tetraédricas, que le confieren una carga global negativa. Sin embargo, esta carga es relativamente débil, razón por la cual no se pueden ligar cationes de una manera fuerte en el espacio interlaminar. Sin embargo e permite la entrada de agua y otras moléculas que causan un aumento en el espacio interlaminar (“hinchamiento” de la arcilla). Con el agua vienen cationes disueltos, que su vez, pueden ser fijados en dicho espacio. Así, las arcillas esmectíticas actúan como depuradoras naturales de los suelos.
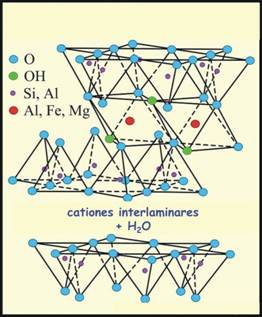
Estructura de las arcillas tipo esmectita. Note el espaciado interlaminar donde se pueden acomodar cationes.
Sin embargo, cuando se supera la capacidad de amortiguación geoquímica del suelo, éste se convierte de hecho en fuente de contaminación. De igual forma, un cambio en las condiciones climáticas puede producir la reversibilidad del proceso. Por ello a menudo se habla de que la presencia de contaminantes en el suelo constituye una bomba de tiempo química, que aún si en un determinado momento no produce efecto alguno, si puede hacerlo en un futuro. El problema radica en que algunos contaminantes se acumulan en los suelos como especies químicas de alta solubilidad. Esto significa que los contaminantes están “disponibles” para que los animales y vegetales que viven en, o sobre el suelo, puedan captarlos, con los consecuentes efectos toxicológicos que ello puede acarrear. El desplazamiento de la contaminación también funciona “hacia abajo”, esto es, las aguas de infiltración pueden transportar los compuestos solubles hacia los acuíferos y contaminarlos. Todo esto nos lleva a decir otros conceptos importantes en lo relativo a la presencia de contaminantes en el suelo: los de geodisponibilidad y biodisponibilidad.
La geodisponibilidad es la consecuencia directa de la actividad minera: al llevar a cabo la explotación minera de un yacimiento, se ponen a disposición del medio geológico unos elementos que antes no estaban, o estaban de una forma mucho más limitada. Cabe destacar, no obstante, que muchos yacimientos minerales, particularmente los de menas sulfuradas, son en sí fuentes naturales de contaminación. Esto depende en gran medida de si son o no aflorantes, de su profundidad (en especial, si se localizan por encima o por debajo del nivel freático), su composición mineralógica, y el clima entre otros.
La biodisponibilidad por su parte, se refiere a la capacidad de un organismo de incorporar metabólicamente un contaminante. Por ejemplo, muchas plantas tienen la capacidad de absorber grandes concentraciones de metales pesados, siempre que se encuentren en el suelo en formas solubles, o asociados a nutrientes básicos. Otras muestran efectos de barrera que hacen imposible la incorporación de un metal pesado pasada una cierta concentración.
Otra labor importante de depuración de soluciones viene dada por otras partículas minerales de los suelos con comportamiento coloidal. Por ejemplo, la goethita (carga positiva en medios ácidos), posee la capacidad para fijar complejos aniónicos arseniacales. Otros óxidos, como los de manganeso constituyen coloides de carga negativa, que les permite fijar cationes en solución.
Suelo rojo, caracterizado por la presencia de fases oxidadas de hierro.
3.6.- Bibliografía
ITGE (1995). Contaminación y depuración de suelos. Publicaciones del ITGE. 330 pg.
Krauskopf, K.B.; Bird, D.K. (1995) Introduction to geochemistry. McGraw-Hill, NY, 647 pp.
McLean, J.E.; Bledsoe, B.E. (1992). Behaviour of metals is soils. USEPA Ground Water Issue, EPA/540/S-92/018.
Porta, J.; López-Acevedo, M.; Roquero, C. (1999) Edafología para la agricultura y el medio ambiente. Ediciones Mundi-Prensa. 849 pg.
SEISnet – Sistema Español de Información de Suelos sobre Internet. http://leu.irnase.csic.es/microlei/microlei2.htm
4.- Prospección geoquímica
Toma de muestras de sedimentos fluviales (Río Toro, Chile) para la determinación de As, Cu, Zn.
Uno de los temas más importantes en geoquímica ambiental es el relacionado con la determinación del grado y extensión de la contaminación. Durante décadas los métodos de prospección geoquímica se utilizaron para detectar yacimientos minerales. Hoy, paradójicamente, estos mismos métodos se empiezan a emplear para determinar el alcance de la contaminación inducida por la actividad minera relacionada con esos mismos yacimientos minerales. La actividad minera genera residuos que se derivan de cuatro fuentes principales: 1) los gases expulsados por las chimeneas de las fundiciones, cuyos compuestos tarde o temprano precipitan en los suelos, a mayor o menor distancia de la fuente de emisión; 2) las escombreras (mineral dumps), con materiales supuestamente estériles pero ricos en minerales altamente reactivos en condiciones atmosféricas, entre estos, sulfuros de hierro, cobre, zinc, plomo, cobalto, mercurio, etc.; 3) de las balsas de estériles (tailings dumps), que similarmente a las escombreras, contienen sulfuros, los cuales a su vez fueron rechazados por el proceso concentrador; y 4) los estanques de solución (pregnant-barren solution ponds) que contienen especies tan nocivas como el cianuro, el ácido sulfúrico, y especies metálica tales como cobre o hierro.

Fuentes potenciales de contaminantes químicos en minería. A: expulsión de gases con SO2 – As2O3 (Chuquicamata, Chile); B: escombrera con minerales de mercurio (Las Cuevas, Almadén); C: balsa de estériles con abundante pirita (Talcuna, Chile); D) estanque con solución cianurada y pila de lixiviación (Punitaqui, Chile).
Todo esto en lo que respecta a la minería, pero esta actividad no es la única que genera residuos solubles, susceptibles de ser movilizados, ya que prácticamente cualquier industria genera un rastro de residuos metálicos u orgánicos que solo en ocasiones son tratados previamente, y en general suelen ir a parar a suelos (y de ahí a las aguas subterráneas), ríos, lagos, o al mar.
¿Cómo detectar la extensión de la contaminación, y la intensidad de la misma? aquí es cuando los viejos métodos de la prospección geoquímica pueden ser de una ayuda inestimable. Dado que el principio básico de la prospección geoquímica se basa en detectar la dispersión de un determinado metal para detectar la fuente de emisión, esto es, el yacimiento mineral, sus principios son válidos también para poder trazar la extensión de un fenómeno de contaminación, ya que las reglas físico-químicas que gobiernan la dispersión de elementos químicos en el ciclo exógeno son las mismas. La dispersión de elementos químicos lleva a la formación de una zona geoquímicamente anómala, que denominaremos anomalía geoquímica. La anomalía geoquímica contrasta claramente con lo que podríamos denominar valores normales de un determinado elemento químico en el medio de dispersión (suelo, aguas, atmósfera). Aunque a veces una anomalía pueda ser obvia en términos numéricos (valores extremadamente altos de un elemento), la caracterización de la misma se lleva a cabo mediante un tratamiento estadístico, más o menos complejo, de la información. Definiremos dos conceptos básicos: 1) valor de fondo (background), que corresponde al valor normal de un elemento en un medio concreto; y 2) anomalía, que corresponde a una desviación estadísticamente significativa a partir del valor de fondo, por ejemplo, los valores que se encuentran sobre dos o tres veces el valor de la desviación estándar (x > 3s). Un concepto relacionado es de umbral (threshold), que corresponde al límite superior de las fluctuaciones de la media, en otras palabras, el límite inferior de los valores anómalos. Es importante tener en cuenta que durante el tratamiento estadístico de los datos geoquímicas puede aparecer más de una población, dando lugar a una población bimodal. En esos casos la segunda población puede ser considerada anómala en su globalidad y el umbral ser fijado en el límite entre ambas poblaciones. En este caso lo más probable es que la segunda población se encuentre relacionada con la anomalía geoquímica (fuente de emisión) que se busca.

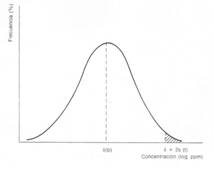
Análisis poblacional. A la izquierda definimos la anomalía en función de los
datos que quedan sobre la media más 2 S; a la derecha tenemos una población bimodal,
los datos pertenecientes a la segunda población pueden ser considerados
como globalmente anómalos.
Por muy sofisticado y discriminante que sea el procedimiento estadístico utilizado para “tamizar” la información, la persona encargada de llevar a cabo el trabajo deberá utilizar además el “sentido común”, ya que cabe recordar que la estadística es tan buena (o mala) como quien la emplea. La utilización de procedimientos estadísticos sin un conocimiento adecuado de la geología de una zona o región puede llevar a errores de gran magnitud. Por ejemplo, la respuesta geoquímica de un elemento químico en una región caracterizada por rocas sedimentarias carbonatadas (e.g. calizas) no es igual a la de una donde las rocas corresponden a secuencias máficas (e.g. basaltos). Tanto los valores de fondo como los halos de dispersión serán diferentes. Así, valores que podrían ser considerados anómalos en la primera no tendrían por qué serlo necesariamente en la segunda.
Existen diferentes escalas a la que se puede llevar a cabo una campaña de prospección geoquímica: 1) de reconocimiento, que tiene un carácter orientativo, basada en mapas 1: 1.000.000 a 1: 500.000; 2) preliminar, donde se utilizan mapas 1: 200.000 a 1: 100.000; y 3) detallada, mapas 1: 50.000 y de mayor detalle. Los conceptos de prospección estratégica y táctica se relacionan con los anteriores, siendo la primera de carácter regional (orientativa), y la segunda de tipo local, sobre blancos (targets) previamente definidos en el estudio regional.
El tipo de muestras que se toman en prospección geoquímica son variadas: rocas, sedimentos fluviales, suelos, aguas, vapores, plantas. La elección depende de múltiples variables, entre ellas: 1) la fisiografía; 2) el marco geológico; 3) el tipo de anomalía que estamos buscando.
Contenidos del resto del Tema:
4.1.- Detectando anomalías geoquímicas mediante la utilización de diferentes tipos de muestras
4.1.1.- Geoquímica de suelos
4.1.2.- Sedimentos fluviales
4.1.3.- Aguas
4.2.- Biogeoquímica
4.3.- Bibliografía
4.1.- Detectando anomalías geoquímicas mediante la utilización de diferentes tipos de muestras
A continuación discutiremos los aspectos generales de la utilización de suelos, sedimentos fluviales, aguas y plantas en prospección geoquímica. Hemos dejado las rocas de lado ya que su utilización tiene más que ver con anomalías primarias (presencia de masas minerales en la roca), que con los problemas de contaminación secundaria inducidas por la actividad industrial.
4.1.1.- Geoquímica de suelos
Las características de los suelos difieren en función de los aspectos geológicos, fisiográficos, y climáticos de una región. Estas condicionan sus perfiles típicos en cuanto al desarrollo (o ausencia) y extensión de los diferentes horizontes (A-C). Bajo el punto de vista de la geoquímica el horizonte B (de acumulación) presenta un gran interés, ya que es ahí donde suelen concentrase de preferencia los elementos químicos.
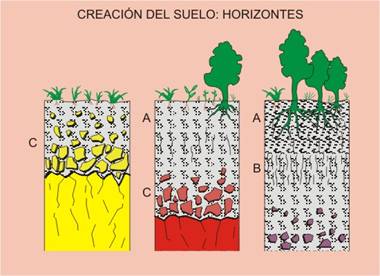
Horizontes del suelo
No obstante, es importante es importante aclarar en este sentido que no existen recetas mágicas, y que por lo tanto conviene estudiar en cada caso cual de los horizontes concentra de preferencia el elemento que estamos investigando. La toma de muestras y estudio del perfil del suelo permite conocer la evolución del contenido metálico en profundidad, y de esta manera posibilita el seleccionar el horizonte más adecuado. Otro problema de interés es el de la distribución de los elementos en las diferentes fracciones granulométricas de los suelos. Aunque generalmente la fracción más fina contiene más minerales de arcilla, materia orgánica, óxidos hidratados de Fe y Mn, y por lo tanto es la más rica en metales, conviene estudiar también cual es la que mejor concentra el elemento investigado. Generalmente se emplea la fracción menor a 80 mallas, lo que no significa que sea necesariamente la mejor en cada caso. El muestreo y análisis de suelos residuales es el método más empleado en prospección geoquímica. Este método de prospección es especialmente útil en las regiones con suelos profundos.
Los estudios orientativos deben constituir el primer paso en la prospección geoquímica de suelos. Dichos estudios buscan determinar la existencia y características de las anomalías asociadas con un determinado foco emisor, y por lo tanto, deben desarrollarse cerca de un lugar con estas características. También deberemos establecer la distribución de elementos en sitios donde positivamente sepamos que no hay contaminación, ya que así podremos determinar de una forma fiables los valores de fondo para cada elemento. También sirven como datos orientativos los valores “normales” (promedio) de los elementos químicos en suelos (Tabla 1).
| Elemento | Suelo y (rango) (ppm) | Plantas (peso en seco) (ppm) |
| S | 700 (30-900) | 3400 |
| Cr | 100 (5-3000) | 0.23 |
| Co | 8 (1-40) | 0.5 |
| Ni | 40 (10-1000) | 3 |
| Cu | 20 (2-100) | 14 |
| Zn | 50 (10-300) | 100 |
| As | 6 (0.1-40) | 0.2 |
| Se | 0.2 (0.01-2) | 0.2 |
| Mo | 2 (0.2-5) | 0.9 |
| Cd | 0.06 (0.01-0.7) | 0.6 |
| Sn | 10 (2-200) | 0.3 |
| Cs | 50 | 0.2 |
| Hg | 0.03 (0.01-0.3) | 0.015 |
| Pb | 10 (2-200) | 2.7 |
Tabla 1: Valores y rangos medios de algunos elementos químicos de interés en temas ambientales.
Datos para suelos y plantas.
Otra labor a desarrollar durante esta etapa inicial es el determinar la naturaleza de la cubierta de suelos, esto es, si trata de una cubierta residual o transportada. También es importante establecer el tipo de análisis que permite definir mejor la zona contaminada. Por ejemplo, más interesante que el contenido total de un metal, puede ser el determinar el contenido de fácil solubilidad, ya que será más indicativo del aporte a través de soluciones ricas en ese metal. Aparte de las fuentes metálicas primarias de origen natural, están los compuestos de origen industrial, que deben ser tomados en cuenta para el análisis final de los datos. Entre estos están los fertilizantes, pesticidas, fungicidas e insecticidas, ya que algunos contienen compuestos metálicos (e.g. Pb(AsO4), Na3AsO3, Cu3(AsO3)2) u organometálicos. La selección del plan de muestreo de suelos se determina principalmente por la forma y dimensión, probada o probable, del foco de emisión. Otro factor a tomar en cuenta es la topografía del terreno, ya que las anomalías geoquímicas se desarrollan pendiente abajo a partir del foco de emisión.
El diseño de la red de toma de muestras tiene que tomar en cuenta las características del problema que queremos tratar (tipo de anomalías buscadas, probable extensión de estas) y el presupuesto con que se cuenta, lo que condicionará el número de muestras que vamos a tomar. El método clásico de muestreo es a lo largo de perfiles, y las características de éstos dependerán entre otras consideraciones de la escala a que estamos trabajando (Tabla 2).
| Escala | Intervalos entre perfiles | Intervalos entre puntos de muestreo |
| 1: 1.000.000 | 12-8 km | 100 m |
| 1: 500.000 | 6-4 km | 100 m |
| 1: 200.000 | 2 km | 100-50 m |
| 1: 100.000 | 1 km | 100-50 m |
| 1: 50.000 | 0.5 km | 50-40 m |
| 1: 25.000 | 250-200 m | 40-20 m |
| 1: 10.000 | 100 m | 20-10 m |
| 1: 5.000 | 50 m | 20-10 m |
| 1: 2.000 | 20 m | 10-5 m |
| 1: 1.000 | 10 m | 5 m |
Tabla 2: características de los perfiles para toma de muestras de suelo dependiendo de la escala de trabajo.
Para el diseño propiamente dicho se determina la dirección que deben tener los perfiles, normalmente perpendicular a la dirección principal que tenga el foco de emisión, si bien se pueden considerar otros factores como el topográfico: perpendicularmente a la pendiente del terreno, si ésta es aproximadamente constante, de forma que podemos verificar que los contaminantes sigan este patrón de migración descendente.
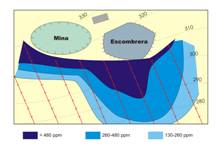
Trazado de perfiles (escala de detalle) para la toma de muestras en función de los focos de contaminación
y la topografía (izquierda). A la derecha se muestra el mapa final que muestra las anomalías
del elemento químico investigado.
Una vez establecida la dirección que deben seguir los perfiles, hay que determinar el espaciado entre éstos y entre muestras sucesivas a lo largo de cada perfil. Una vez establecidos estos parámetros, se pasa al trabajo de campo, cuyo primer paso es la localización de los puntos de muestreo previstos en la malla diseñada. Estos puntos se localizan adecuadamente mediante taquimetría (o brújula) y cinta métrica, señalizándose mediante estacas de madera o metal, y se procede a la toma de muestras, mediante métodos “artesanales” (azada, pico, pala) o mediante herramientas diseñadas al efecto.

Toma de muestras de suelo en el distrito de Almadén (España) para la
determinación de contenidos de mercurio.
Una vez tomadas las muestras, se pasa a su preparación para el análisis. Las muestras de suelos requieren un secado como primer paso. Para ello se extienden sobre papel de filtro o similar, en lugares cálidos, secos y bien aireados, pero evitando excesiva corriente de aire que pueda levantar polvo y mezclar las muestras. Una vez secas las muestras, se suele proceder a su cuarteo, para obtener distintas fracciones, que se emplearán para diferentes determinaciones. Para ello se emplean cuarteadores específicamente diseñados, o el método manual, basado en homogeneizar la muestra lo más posible, hacer con ella un “pastel” y dividirlo en cuatro cuartos. Se toman los cuartos opuestos por el vértice para una fracción y los otros dos para otra, sucesivamente hasta obtener la o las muestras que se requieran. Durante este proceso es conveniente deshacer los grumos de muestra apelmazados por la materia orgánica, para lo cual suele ser muy útil un rodillo de amasar.


Operaciones de preparación de muestras en el laboratorio:
de cuarteo (izquierda) y de deshacer “grumos” (derecha).
También es conveniente eliminar los cantos de 1 cm o superior, que por lo general no suelen tener interés para el estudio. Una vez obtenidas estas fracciones o alícuotas de la muestra total, se reserva una para posibles comprobaciones (e.g. mediante el envío de algunas muestras a otro laboratorio), y el resto se dedican a las determinaciones requeridas. A menudo durante la preparación de la muestra se contempla su tamizado, ya que como hemos visto, muchas determinaciones químicas se hacen sobre muestras de tamaño de grano específico ya que los elementos a identificar se concentran en determinadas fracciones granulométricas, sobre todo en la fracción arcilla (metales pesados). Para ello se emplean unidades de tamizado adecuadas a la granulometría requerida, que es necesario conservar limpias entre muestra y muestra para evitar la contaminación de muestras sucesivas. En otros casos hay que proceder a la molienda de las muestras. Cuando los elementos de interés pueden estar concentrados en las fracciones más gruesas, se procede a esta molienda para homogeneizar el tamaño de partículas que se envían al laboratorio. En este caso es muy conveniente conocer la composición química del molino o mortero por el o por los que pasan las muestras, para evitar problemas de contaminación. En este sentido es vital la limpieza absoluta del equipo (mediante aire a presión y/o agua, o acetona, para evitar procesos de oxidación) entre muestras sucesivas, para evitar su contaminación. En definitiva, la preparación de las muestras es un proceso físico, en el que es posible que se produzcan problemas de contaminación, que solamente podrán evitarse siendo extremadamente cuidadosos en la limpieza de los equipos a utilizar y en un control detallado de cada uno de los procesos que llevemos a cabo.
4.1.2.- Sedimentos fluviales
Los sedimentos clásticos están compuestos principalmente por los productos menos solubles de la meteorización. También pueden incluir elementos móviles como parte de los materiales clásticos y del material removido de las aguas y adsorbido en los sedimentos (metales adsorbidos en minerales de arcillas u oxihidróxidos de Fe o Mn). Una medida del material removido de las aguas viene dado por la razón: metal de fácil extracción química/metal total. Esta razón puede llegar hasta el 50 % en áreas de de rápida precipitación del metal en solución, generalmente cerca de las fuentes de emisión.
El contraste entre los valores anómalos y el valor de fondo depende de varios factores, entre ellos: 1) del contraste original en los materiales desde donde provienen los sedimentos; de la fracción de sedimento analizada; y 3) del método de análisis. A su vez, la persistencia del contenido metálico de los sedimentos al alejarnos de la fuente de emisión depende de: 1) del aporte de metal a lo largo del río desde otras posibles fuentes; y 2) de la mezcla con sedimentos de bajo contenido metálico. Estas dos a su vez se relacionan con períodos estacionales y fisiográficos; en épocas de lluvias hay mayor capacidad de transporte y mayor aporte desde las zonas más fácilmente erosionables.
Al contrario de las aguas de un río, cuyo contenido en elementos es más bien homogéneo (sujeto eso sí a cambios en el caudal), los sedimentos presentan heterogeneidades que dependen de: 1) la distribución de los puntos de entrada del metal en el cauce; 2) los fenómenos de especiación del metal; y 3) la variación en el tipo de sedimento a lo largo del río. Las muestras de sedimentos tienden a ser más homogéneas cuando el metal se encuentra adsorbido a la fracción fina, y las variaciones sedimentológicas se deben a cambios locales de velocidad y caudal de los ríos.
Las características que debe tener el muestreo de sedimentos fluviales en función de la escala de trabajo se muestra en la tabla 3.
| Escala | Longitud mínima del cauce a muestrear | Número de puntos de muestreo por km2 |
| 1: 200.000 | 0.8 km | 1.7-2.1 |
| 1: 100.000 | 0.4 km | 4.0-5.0 |
| 1: 50.000 | 0.2 km | 8.5-14.0 |
| 1: 25.000 | 0.1 km | 18.0-32.0 |
Tabla 3: Características recomendadas del muestreo de sedimentos fluviales en función de la escala de trabajo.
Otro aspecto a tener en cuenta es la naturaleza de las unidades de roca que corta el río. Determinados tipos litológicos pueden ser ricos o extremadamente ricos en un determinado metal, y por lo tanto dar lugar a falsas anomalías. Por ejemplo, si estamos estudiando cromo, y el río en un sector corta rocas de composición peridotítica, es posible que obtengamos una anomalía basada solamente en el valor de fondo de estas rocas, que no tenga nada que ver con la fuente industrial del metal. Esto nos lleva a recordar la importancia de los estudios geológicos “previos” al diseño de una campaña de prospección geoquímica. El clima y la fisiografía son factores importantes a considerar ya que pueden afectar seriamente la movilidad de un determinado elemento. La alta movilidad geoquímica del Zn se ve reducida en regiones montañosas y frías. A pesar de que parece en principio una contradicción, la prospección geoquímica de sedimentos fluviales puede ser de gran interés en regiones áridas y semiáridas, ya que aunque esporádicas, las precipitaciones pueden ser muy intensas, lo cual posibilita, en ocasiones, fuertes tasas de remoción del regolito, y por lo tanto, una fuerte movilización de los elementos químicos ligados a éste.
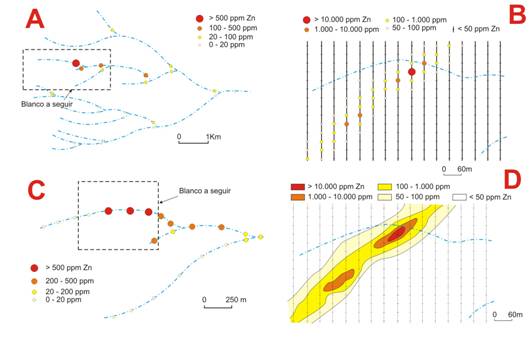
Toma de muestras de sedimentos fluviales a distintas escalas (A-C) para la definición de un blanco (B-C),
y su posterior caracterización de detalle (D).
Cabe destacar que cuando sea posible, conviene combinar estos estudios con los de aguas. La toma de muestras debe de ir acompañada de la recogida de datos sobre las características físico-químicas de las aguas, concentración de oxígeno, pH, etc.


A la izquierda, toma de muestras de sedimentos fluviales (Río Toro, Chile)
para la determinación de As, Cu, Zn. A la derecha laboratorio de campo
para la determinación del pH y contenido de oxígeno de las aguas.
En cuanto a la preparación de muestras, esta se efectúa de una manera equivalente a la anteriormente descrita para las muestras de suelo.
4.1.3.- Aguas
Se denomina anomalías hidrogeoquímicas a los patrones anómalos presentes en las aguas superficiales y subterráneas. La importancia de la migración de los elementos químicos y complejos en el agua está controlada por movilidad geoquímica (Tabla 4).
| Movilidad relativa | Condiciones de Eh-pH | ||
| Oxidante,
pH> 4 |
Reductor, ausencia de H2S | Reductor,
Con H2S |
|
| Muy móvil | S, Cl, Br, I, B | Cl, Br, I, B, Rn | Cl, Br, I, B, Rn |
| Móvil | Ca, Na, Mg, F, Sr, Zn, U, Mo, V, Se, Te, Re | Ca, Na, Mg, F, Sr, Mn2+, Fe2+, Zn, Cu, Ni, Pb, Cd | Ca, Na, Mg, F, Sr |
| Ligeramente móvil | Si, K, Mn, P, Ba, Li, Rb, Cs, Pb, Ni, Cu, Co, As, Cd, Tl, Hg | Co, Hg, Ag, Si, K, P, Ba, Li, Rb, Cs, As, Tl, Ra, Hg, Ag | Si, K, P, Ba, Li, Rb, Cs, Tl, Ra |
| Inmóvil | Fe, Al, Ga, Cr, Ti, Zr, Hf, Y, REE, Nb, Ta, Be, Th, Sn, PGE, Au | S, V, Mo, Se, Te, U, Re, Mn, Cu, Ni, Pb, Cd, Fe, Co, Hg, Ag | |
Tabla 4: Movilidades relativas de diferentes elementos químicos en función del Eh y pH.
La aplicabilidad de la hidrogeoquímica a los elementos que se encuentran en baja concentración depende de los medios analíticos de que se dispone. Cabe hacer notar que la mayoría de los metales se encuentran en concentraciones muy bajas en las aguas naturales, en el orden de las partes por billón o partes por trillón (micro o nanogramos por litro). La persistencia de las anomalías hidrogeoquímicas es un factor determinante y está condicionado por: 1) el contraste inicial; 2) la dilución; 3) la precipitación. El contraste inicial depende de la solubilidad de los elementos químicos en la fuente, la fisiografía y el clima. La dilución se debe al efecto de las aguas de afluentes con contenidos metálicos más bajos. Por último, la precipitación de los elementos está condicionada por las fases minerales de los sedimentos, el Eh, y el pH. Muchos metales divalentes (e.g. Cu, Zn, Ni, Co, Cd, Pb) y trivalentes (Fe, Al) muestran correlación una inversa con el pH, es decir su solubilidad aumenta a medida que disminuye éste. El aumento del Eh también puede condicionar la precipitación de los cationes, así como ayudar a la formación de oxihidróxidos de Fe (e.g. goethita: FeO(OH)) que pueden substraer metales del agua (e.g. As, Pb). Por otra parte las condiciones oxidante pueden incrementar la movilidad de un elemento, como en el caso del uranio (ión uranilo: UO22+), el cual precipita en condiciones reductoras.
Las anomalías hidrogeoquímicas dependen mucho de las variaciones climáticas (precipitaciones, temperatura) y por lo tanto de los cambios estacionales. De ahí que los resultados que podamos obtener de las aguas de un río en verano e invierno puedan diferir fuertemente. La lluvia genera un aumento del caudal y por ende de la dilución. Sin embargo, si esto sucede después de un período seco las aguas podrán lixiviar materiales ricos y transportar los metales, generando de esta manera una elevación en la concentración de estos. Esto puede ser notable en regiones semiáridas del planeta, y uno de los ejemplos más notables vienen dado por las inundaciones del año 1997 en el Río Carson (Nevada, USA), las que ayudaron a remover y poner en solución grandes cantidades de mercurio presentes en antiguas escombreras abandonadas (hasta 61 ppm Hg en las aguas).
Las muestras de agua se guardan en botellas o botes de polietileno de un cuarto o medio litro (dependiendo del número de elementos a analizar). Con el objeto de evitar posteriores oxidaciones del Fe2+ disuelto que pudieran originar su precipitación y la consecuente remoción de ortos metales, es conveniente llenar el recipiente, y si es posible, acidularlo ligeramente. El análisis de las muestras se puede realizar también en el campo, aunque con algunas obvias limitaciones. Aunque los métodos de campo son menos precisos y consumen tiempo de trabajo tienen la ventaja de que no hay que transportar las muestras y que se pueden tomar decisiones inmediatas.
4.2.- Biogeoquímica
El uso de la vegetación como método de prospección de prospección involucra la respuesta de las plantas a su medio, en particular al substrato químico que las soporta. Esta metodología se basa en el análisis químico de las plantas (Tabla 1) como medio para obtener evidencias acerca de las posibles anomalías geoquímicas que se oculten en profundidad. La biogeoquímica se adapta muy bien a aquellas regiones que presentan una vegetación muy densa y donde la cartografía geológica es difícil de ser llevada a cabo (ausencia de afloramientos). Aunque esta técnica ha probado ser de indudable ayuda, también presenta sus limitaciones, ejemplificadas en el denominado efecto barrera: con algunas pocas excepciones, las plantas pueden acumular un determinado elemento hasta cierto nivel solamente. De hecho, en el caso del mercurio, las raíces pueden actuar a modo de barrera impidiendo que el elemento ascienda hacia los órganos superiores de la planta. En este sentido la plantas pueden ser clasificadas en cuatro categorías: 1) sin efecto de barrera, las que concentran linealmente elemento químico investigado; 2) semi-barrera, que concentran entre 30 y 300 veces el valor de fondo del elemento en la planta; 3) con barrera, contenidos de hasta 3-30 veces el valor de fondo; y 4) con barreara de fondo, que no superan las concentraciones normales del elemento en una determinada planta. En otra esquema de clasificación, se habla de plantas hiperacumuladoras cuando determinadas especies toleran 10-100 veces más los valores normales de un determinado elemento (Tabla 5).
| Elemento | Especie | Contenido normal (ppm) | Contenido máximo (ppm) | Localidad |
| Cu | Becium homblei | 183 | 2.500 | Zambia |
| Mn | Fucus vesiculosus | 4.815 | 90.000 | Rusia |
| Ni | Alyssum Bertolonii | 65 | 100.000 | Italia |
| Zn | Thlaspi calaminare | 1.400 | 10.000 | Alemania |
| Zn | Thlaspi caerulescens | 43.710 | Europa central | |
| Cd | Thlaspi caerulescens | 2.130 | Europa central | |
| Pb | Thlaspi caerulescens | 2.740 | Europa central |
Tabla 5: Algunas plantas hiperacumuladoras y sus contenidos en metales pesados.
Existen informes de 1885 que ya mostraban que Thlaspi calaminare acumulaba Zn en grandes concentraciones, en cuanto a Ni el informe más antiguo que hace mención a esta capacidad de las plantas data de 1948, y trató sobre el caso de Alyssum bertolonii, que podía contener hasta más de 1 % (> 10.000 ppm) de Ni en su peso en seco.

Plantas hiperacumuladoras: A: Thlaspi caerulescens (Zn y Ni); B: Alyssum bertolonii, (Ni);
C: Euphorbia helenae (Ni); D: Justicia lanstyakii (Ni).
Los factores determinantes de anomalías de origen químico en las plantas son: 1) la disponibilidad de elementos en el suelo; 2) las características nutricionales de la planta; y 3) los factores químicos y biológicos de la incorporación de elementos químicos en la planta. La disponibilidad de elementos químicos en el suelo depende tanto de la concentración del elemento como de su movilidad respecto de la actividad de la planta. Esto es a su vez función del pH, Eh, la capacidad de cambio de bases, y la presencia de agentes acomplejantes. Las raíces, además de captar las sales disueltas pueden absorber material unido a la superficie de partículas clásticas, en parte debido al efecto de microambientes ácidos generados por la raíces y en parte por fenómenos de cambio de bases.
Absorción de Ni desde un suelo enriquecido en el elemento
El movimiento de constituyentes inorgánicos a la planta es controlado selectivamente, de manera que algunos elementos son admitidos libremente mientras que otros son rechazados en mayor o menor grado. Como hemos comentado anteriormente, aunque las raíces rechacen (en mayor o menor grado) selectivamente a algunos elementos tales como el plomo, el vanadio o el mercurio, una parte significativa llega a los órganos superiores de la planta y puede ser fácilmente detectada mediante análisis químicos.
Los principales elementos químicos requeridos por las plantas son N, K, P, S, Ca, y Mg. Además de éstos los vegetales requieren muchos elementos traza, principalmente Cu, Zn, Mo, Mn, o B. Si los suelos no los poseen de manera biodisponible y en la cantidad adecuada, las plantas sufren trastornos fisiológicos o mueren. Por otra parte, un exceso de estos elementos sobre un nivel crítico es también perjudicial para la vida de la planta (biotoxicidad). En muchas especies de plantas el rango entre el mínimo requerido y el máximo tolerado es estrecho, por ejemplo, unas pocas ppm en el caso del boro, mientras que elementos como el cobre y el zinc pueden ser tolerados en mayores cantidades.
Un factor de mucho interés en los estudios de prospección biogeoquímica es la profundidad de penetración de las raíces. Aquellas plantas de raíces profundas que obtienen el agua del nivel freático se denominan freatófitas, mientras que aquellas de raíces extendidas en las capas superficiales del suelo son denominadas xerófitas. Las primeras son de especial interés ya que permiten detectar contenidos metálicos a profundidades notables, de 10 a 15 m e incluso más.
La toma de muestras puede realizarse sistemáticamente (red de muestreo) o bien tomando muestras de plantas donde sea posible (si la especie investigada es poco abundante). Una muestra de 20 g de materia vegetal es suficiente para dar 1 g de cenizas requerido para el análisis químico. Aunque potencialmente cualquier parte de la planta es susceptible de ser analizada, las hojas constituyen un blanco común dado su fácil acceso durante un muestreo sistemático. De todas maneras es muy conveniente probar en un estudio preliminar que parte de la planta concentra mejor el elemento investigado.
Esta capacidad de algunas plantas de acumular elementos químicos las hace doblemente interesantes, ya que por un lado, como hemos visto, nos permiten detectar anomalías en un determinado elemento químico, y por otra, dada su capacidad de acumulación, pueden ser utilizadas para “limpiar” un terreno contaminado.
4.3.- Bibliografía
Chaney, R., Brown, S., Li, Y.M., Angle, J.S., Homer, F., y Green, C. 1995. Potential use of metal hyperaccumulators. Mining Environmental Management, 3: 9-11.
Gammon, J.B. 1988. Gold !!! and other metals: how they are found and mined. IMM, London, 83 pp.
Levinson, A.A. 1980. Introduction to exploration geochemistry. Applied Publ. Ltd. Ill., 924 pp.
Oyarzun, R. 1991. Prospección geoquímica: I) Conceptos básicos. En: Yacimientos Minerales, Lunar, R. & Oyarzun, R. (eds.) 1991. Editorial Centro de Estudios Ramón Areces, Madrid, 811-818.
Oyarzun, J., y Oyarzun, R. 1991. Prospección geoquímica: III) Geoquímica de suelos, sedimentos fluviales, aguas, biogeoquímica y geobotánica. En: Yacimientos Minerales, Lunar, R. & Oyarzun, R. (eds.) 1991. Editorial Centro de Estudios Ramón Areces, Madrid, 837-856.
Reeves, R.D., Baker, A.J.M., y Brooks, R.R. 1995. Abnormal accumulations of trace metals by plants. Mining Environmental Management, 3: 4-8.
Siegel, F.R. 1974. Applied geochemistry, John Wiley & Sons, NY, 353 pp.
5.- Mineralogía y Residuos mineros

Drenaje ácido proveniente de las escombreras en la mina abandonada de San Quintín (Ciudad Real, España)
Durante los procesos mineros se originan importantes residuos de origen y composición muy variables. Estos van desde el polvo (particulado fino) que se origina durante las labores de explotación, pasando por los efluentes líquidos que se generan durante el proceso minero (la propia mina, los lavaderos, las escombreras), hasta los residuos sólidos que se acumulan en las escombreras, y claro está, los gases liberados por los procesos mineros y metalúrgicos. Cada uno de estos residuos presenta problemas ambientales importantes, que describiremos en el presente tema.
Organización del Capítulo:
5.1.- Emisiones a la atmósfera
5.1.1.- Polvo
5.1.2.- Gases
5.2.- Efluentes líquidos
5.3.- Residuos sólidos
5.4.- Bibliografía
5.1.- Emisiones a la atmósfera
Una de las principales emisiones que genera la minería a la atmósfera es el polvo. Otro aspecto a considerar es que determinados minerales al descomponerse o al ser sometidos a procesos metalúrgicos dan origen a la emisión de gases de naturaleza variada.
5.1.1.- El polvo
Como veremos con más detalle en el tema de minerales y salud humana, las características del polvo que afectan a ésta son fundamentalmente la composición y granulometría. La composición afecta porque determinados minerales contienen metales que a su vez pueden tener efectos tóxicos. Por otra parte, la granulometría es muy importante puesto que las partículas de polvo de tamaño inferior a 10 micras (las denominadas PM10) entran en el sistema respiratorio alcanzando los pulmones, donde pueden quedar acumuladas y generar graves daños al sistema respiratorio (e.g. silicosis). Las partículas menores de 2.5 micras (denominadas PM2.5) son aun más peligrosas, ya que se mantienen en suspensión en al aire, lo que permite que se desplacen muy largas distancias.
El polvo generado por los procesos mineros puede proceder de distintas fuentes:
– Voladura y arranque. Es una fuente muy importante de este contaminante, puesto que normalmente se lleva a cabo sobre roca seca, lo que facilita aún más el desprendimiento del polvo.


Emisiones polvo causadas por voladura de roca en minas a cielo abierto
– Carga. En este caso es más sencillo de evitar o controlar, puesto que durante el proceso se puede proceder al regado del material.

Operaciones de carga de mineral. Mina de cobre Zaldivar (Chile).
– Transporte. Si se realiza mediante cinta transportadora el viento podrá levantar y arrastrar un volumen significativo de polvo, si no se evita mediante el humedecimiento del material transportado, o utilizando cintas cerradas. Si el transporte es en vehículos, se puede producir una doble emisión e polvo: la relacionada con el escape de polvo procedente de la propia carga, y el polvo levantado durante la rodadura del vehículo. Ambos son relativamente sencillos de controlar, en un caso recubriendo la carga, en el otro regando las pistas de rodadura.

Polvo levantado por un camión durante el transporte de minerales.
– Molienda. Es otra de las fuentes más importantes de este elemento, puesto que como el arranque (voladura de roca) por lo general se realiza en seco. Por otra parte, se puede llevar a cabo dentro de edificaciones realizadas al efecto, lo que permite evitar la dispersión generalizada del polvo que se genera.

Operaciones de molienda primaria.
– Disposición en escombreras. El volcado del estéril (u otros productos) en escombreras produce una emisión inmediata, durante el propio proceso de descarga, y un riesgo potencial de emisión, si la escombrera no se trata de alguna forma para prevenir que el viento levante polvo de la misma.

Descarga de minerales.
– Metalurgia. El tratamiento metalúrgico de menas puede llevarse a cabo de distintas maneras, siendo las más comunes la pirometalugia (descomposición térmica de los minerales o tostación) y la hidrometalurgia (descomposición del mineral en medio acuoso, mediante reactivos específicos que permiten la separación del elemento de interés minero). En el primer caso se suelen emitir a la atmósfera gases y partículas, mientras que en el segundo caso las emisiones pueden ser de aerosoles que contienen reactivos más o menos tóxicos (e.g. cianuro, ácido sulfúrico), y se puede levantar polvo de las pilas de lixiviación. El particulado emitido por las chimeneas puede ser muy importante en algunos casos, como en Chuquicamata (ver siguiente figura) donde por ejemplo el año 2002 se emitieron unas 1040 t toneladas de este tipo de material.

Pirometalurgia, fundición de cobre de Chuquicamata (Chile).

Hidrometalurgia: cianuración en pila en la mina El Soldado (Chile).
En cuanto a la naturaleza del polvo generado, puede ser de distintas tipologías:
– Polvo mineral. Es el más común, y puede ser muy variado en sus características: su composición dependerá de la del material minero explotado, mientras que su granulometría dependerá por una parte de la composición, y por otra del proceso minero que lo genere: será más grueso el levantado durante las voladuras, y más fino el procedente de procesos de molienda y subsecuentes.

Fragmentos de cuarzo fácilmente inhalables (imagen SEM).
– Polvo de rodadura. Dependerá de la naturaleza del firme de rodadura sobre el que se muevan los camiones.
– Cenizas. Son las partículas procedentes de procesos de pirometalurgia o de combustión de productos mineros, como el carbón. También podrán tener composiciones variadas, en función de la naturaleza del proceso y de los productos afectados por el mismo. La granulometría será por lo general muy fina, muy inferior a la característica de los procesos mineros propiamente dichos.

Emisiones de humo con cenizas desde una planta generadora de electricidad por combustión de carbones.
Los minerales que podemos encontrar en forma de partículas en el aire, pueden ser muy variados: los más comunes son el cuarzo, por su gran abundancia y resistencia a la alteración meteórica, y las arcillas, que son también muy comunes en medio exógeno, y que por su morfología plana y su pequeño tamaño de grano suelen constituir la fracción más fina del polvo en suspensión.
5.1.2.- Los gases
Durante los procesos mineros y afines puede producirse la emisión de gases, bien porque estén contenidos en aquello que explotemos (caso del grisú en el carbón), o porque se produzca una descomposición del mineral que libere esos gases durante el proceso metalúrgico. En concreto podemos citar los siguientes casos como más significativos:
– El grisú es un gas altamente explosivo que se libera durante la explotación del carbón y que esta formado por una mezcla de tres gases: metano, nitrógeno y anhídrido carbónico. Para que se verifique la explosión de grisú se necesita que se cumplan tres condiciones: a) Que la relación del metano al aire este en proporción determinada (5% al 15%); que a esta mezcla apropiada se le aplique una fuente de calor con temperatura mínima de 650º C; y c) que dicha llama de ignición permanezca durante un tiempo determinado en contacto con la mezcla grisuosa, lo que se conoce con el nombre de “retardo a la inflamación” o “histéresis de encendido”, propiedad en que se fundan los explosivos de seguridad. Con llama a 650ºC el “retardo” se aproxima a 10 segundos, mientras que si la temperatura alcanzara los 1000ºC, el “retardo” solo sería de 1 segundo.

Mina de carbón subterránea.
La pirometalurgia libera una gran cantidad de gases, entre los cuales suelen destacar los óxidos de azufre, puesto que esta técnica se aplica a los sulfuros. Por ejemplo, los procesos que ocurren dentro de un horno convertidor de cobre (producción de cobre blister) son del tipo:
2 Cu2S + 3 O2 → 2 Cu2O + 2 SO2
2 Cu2O + Cu2S → 6 Cu + SO2
un proceso que libera SO2 a la atmósfera.

Convertidores para la fundición de sulfuros y producción de cobre metálico.
– Otros gases ligados a la pirometalurgia pueden ser muy variados: vapor de mercurio en el caso de la tostación de cinabrio o de esfalerita u otras menas ricas en este metal, vapores de arsénico o de cadmio en la tostación de menas de cobre o de cinc, etc.

Parte del complejo metalúrgico de Almadén (España) para la tostación de cinabrio (HgS).
Parte del material escapa a la atmósfera como Hg0 gaseoso.
– Vapores de flúor liberados por los minerales de la arcilla durante la cocción de los productos de alfarería o cerámica en general.
La problemática de estas emisiones normalmente no se puede resolver más que mediante aplicación de tecnologías industriales a los correspondientes procesos. Un caso notable en este sentido eran por ejemplo las emisiones de arsénico de la fundición de Chuquicamata (Chile), con valores de 2340 toneladas/año en 1994. En la actualidad la compañía minera CODELCO tiene que recuperar al menos una parte importante del arsénico que potencialmente sería emitido. En Chuquicamata el proceso se realiza en una planta hidrometalúrgica que recupera el cobre, y precipita el arsénico como arsenato férrico. A partir del 2003 las cifras de emisión no podrán exceder las 400 toneladas/año.
5.2.- Efluentes líquidos
Los efluentes líquidos mineros tienen su origen en la manipulación de los productos mineros con agua o soluciones químicas. Por ejemplo, durante los procesos de concentración por vía húmeda o por el empleo del agua para lavar instalaciones mineras. A esto tenemos que sumar las interacciones naturales que se produce entre los productos mineros y las aguas superficiales o de lluvia. Por ejemplo la lluvia cae sobre las escombreras, infiltrándose en las mismas, generado procesos de oxidación, hidrólisis, lavado, etc.

Drenaje ácido proveniente de las escombreras en la mina abandonada de Pb-Zn San Quintín (Ciudad Real, España)
El contacto entre los minerales y el agua puede originar distintas reacciones, en función de la naturaleza del mineral o minerales implicados y de la físico-química del agua implicada. En cuanto a la mineralogía implicada, cada mineral presenta distintos comportamientos frente al agua: los hay solubles e insolubles, hidrolizables y no hidrolizables, sorbentes y no sorbentes. Los minerales solubles a su vez pueden serlo en diferentes grados, y depender de la temperatura del agua. La halita y el yeso son ejemplos de minerales solubles en distinto grado. La halita es muy soluble incluso en agua fría mientras que el yeso es mucho menos soluble, aunque su solubilidad aumenta considerablemente con un incremento en la temperatura.
La hidrólisis de los minerales consiste en su descomposición debida a la acción de los hidrogeniones de las aguas ácidas. El proceso implica tres pasos: 1) rotura de la estructura del mineral. Debido a su pequeño tamaño y a su gran movilidad, los iones H+ se introducen con facilidad en las redes cristalinas, lo que produce la pérdida de su neutralidad eléctrica; para recuperarla, el cristal tiende a expulsar a los cationes, cuya carga es también positiva. Como consecuencia, la estructura cristalina colapsa, y se liberan también los aniones. 2) Lixiviado de una parte de los iones liberados, que son transportados por las aguas fuera de la roca meteorizada. 3) Neoformación de otros minerales, por la unión de los iones que dan como resultado compuestos insolubles. La intensidad del proceso hidrolítico se traduce en el grado de lixiviación de elementos químicos y en la formación de nuevos minerales.
El drenaje ácido que se produce por la oxidación e hidrólisis de los sulfuros, y en especial de la pirita, puede ser resumido de la siguiente forma:
4 FeS2 + 14 O2 + 4 H2O → 4 Fe2+ + 8 SO42- + 8 H+
a su vez, los iones ferroso (Fe2+) se oxidarán de la siguiente manera:
4 Fe2+ + O2 + 4 H+ → 4 Fe3+ + 2 H2O
y los iones férricos se hidrolizan para formar goethita:
Fe3+ + 2 H2O → 4 FeO(OH) + 3 H+
Durante el transporte de los productos de la meteorización el agua arrastra tres componentes mineralógicos: la carga sólida (partículas en suspensión, que corresponden realmente a minerales), coloides (que corresponden a precursores de minerales arcillosos u oxidados, que pueden flocular a partir de este componente), y iones en disolución, que pueden precipitar en forma de compuestos minerales si cambian las condiciones físico-químicas del agua portadora.
La carga sólida, por tanto, serán granos minerales de naturaleza variada, aunque siempre con una claro predominio de minerales de la arcilla, tanto por ser minerales muy abundantes como productos de la meteorización, como porque su pequeño tamaño de grano favorece su transporte en suspensión. Al ser minerales que pueden presentar propiedades de sorción, a menudo estas partículas arcillosas actúan como colectores de contaminantes que el agua arrastra en disolución: determinados iones son capaces de ser fijados por estas partículas, con lo que al depositarse incorporan estos iones al sedimento correspondiente. Una vez en el mismo, la físico-química de las aguas puede favorecer la retención o movilidad de estos iones. Los parámetros que regulan el sistema son: la salinidad, el potencial redox (Eh), y el pH: un incremento de la salinidad conlleva una competencia, entre metales pesados y metales grupos I y II, por los sitios de ligazón (p.ej., espaciado interlaminar en las arcillas), lo que se traduce en la expulsión de los metales pesados, y su devolución a la columna de agua; un incremento del Eh genera la inestabilidad de los compuestos reducidos (e.g., sulfuros), poniendo el paso del metal a la disolución; un decrecimiento del pH tiene dos efectos: 1) induce la disolución de otros sulfuros; y 2) aumenta la solubilidad de los metales disueltos. Por ejemplo:
CuFeS2 + 16Fe3+ + 8 H2O → Cu2+ + 2 SO42- + 17 Fe2+
2 PbS + 4 Fe3+ +3 O2 + 2 H2O → 2 PbSO4 + 4 Fe2+ + 4 H+
2 FeAsS + Fe2(SO4)3 → 2 H3AsO4 + 4 FeSO4 + H2SO4
Cu3AsS4 + 5.5 Fe2(SO4)3 + 4 H2O → 3 CuSO4 + 11 FeSO4 + 4 S + H3AsO4 + 2.5 H2SO4
Zn1-xCdxS + 8 Fe3+ + 4 H2O → 8 H+ + (1-x) Zn2+ + (x) Cd2+ + SO42- + 14 Fe2+
Los coloides transportados por el agua pueden flocular cuando las condiciones del agua lo permiten, y durante el proceso pueden también arrastrar al sedimento otros componentes disueltos del agua. Especialmente interesantes desde el punto de vista de lo que ocurre en el entorno minero es el comportamiento de los geles de óxidos-hidróxidos de hierro. Los iones que el agua transporta tienen diferentes tendencias a la formación de compuestos minerales, insolubles, en función de la presencia de otros iones, y de la físico-química del agua portadora.
5.3.- Residuos sólidos
Los principales residuos sólidos generados por la minería son las escombreras.

Escombrera de minerales abandonada en Morrón de Mateo (Almería, España).
Desde el punto de vista mineralógico, cada escombrera tendrá unas características peculiares, en función de los siguientes parámetros:
– Naturaleza de la explotación.
– Minerales acompañantes: a menudo los minerales metálicos de la ganga, que por definición van a parar a las escombreras, representan el máximo riesgo medioambiental: es el caso de la pirita, que podemos encontrar en cualquier tipo de yacimientos metálicos y en los de carbón, y que genera los problemas a que ya nos hemos referido.
– Granulometría: el tamaño de grano de los componentes de la escombrera condiciona la superficie reactiva de los granos, con lo cual las escombreras formadas por granos menores son más reactivas.
– Climatología: los climas cálidos y semiáridos favorecen las reacciones de hidrólisis, generadoras de los procesos antes descritos.

Derrame en la selva tropical de un concentrado de sulfuros de cobre,
el cual se ha oxidado (OK Tedi, Papua Nueva Guinea).
– Acción bacteriana: las bacterias catalizan de forma muy importante el desarrollo de muchas de las reacciones antes descritas, puesto que obtienen energía de su acción. Por ejemplo, Por ejemplo, la bacteria Thiobacillus ferrooxidans es la mayor responsable de la contaminación relacionada con el drenaje ácido procedente de explotaciones mineras y mineralizaciones en general. Esta es una bacteria acidófila (propia de ambiente ácido), con una fisiología basada en la fijación de carbono a partir del CO2 atmosférico, obteniendo su energía a partir de oxidación del hierro o azufre:
Fe2+ + 0.25 O2 + H+ → Fe3+ + 0.5 H2O
H2S + 2 O2 → SO42- + 2 H+
Bacterias T. ferrooxidans (flecha) adheridas a una partícula de sulfuro.
[/vc_column_text][/vc_tta_section][vc_tta_section title=»6.- Almacenamiento de residuos» tab_id=»1531830999743-342bd493-8704″][vc_column_text]
Redistribución de residuos en un vertedero.
Organización del Capítulo:
6.1.- Residuos: tipología
6.2.- El entorno adecuado
6.3.- El empleo de minerales y rocas en el propio almacén
6.4.- Los almacenes de residuos nucleares: Almacenes Geológicos Profundos
6.4.1.- Formación geológica albergante
6.4.2.- Barreras de ingeniería
6.5.- Bibliografía
6.1.- Residuos: tipología
La humanidad genera volúmenes inmensos y crecientes de residuos de todo tipo, cuya gestión se está transformando en uno de los mayores problemas que tiene planteada la sociedad moderna. En este ámbito, la conciencia conservacionista surgida a partir de los años 70 ha hecho surgir normativas muy diversas, basadas fundamentalmente en el reciclado y reutilización de los materiales ya utilizados. Sin embargo, un volumen muy significativo de residuos no puede ser reciclado ni reutilizado por motivos diversos: pueden ser materiales para los que no se encuentren usos adecuados en ese momento, o cuyo empleo represente un riesgo para la salud o para el medio ambiente. Estos últimos son los denominados “residuos tóxicos y peligrosos”.


Iconografía que denota residuos peligrosos.
A la izquierda biológicos, a la derecha radioactivos
En el ámbito legislativo relativo a residuos tóxicos y peligrosos, en España se ha dado en los últimos años un proceso similar al que se había producido con anterioridad en los países más avanzados. Hasta mediados de la década de los 80, no se ha contado con la legislación específica sobre esta materia. La Ley sobre “Desechos y Residuos Sólidos Urbanos” de 1975 (BOE, 1975) fue promulgada en el mismo año que la CEE dictaba su Directiva 75/442/CEE relativa a la gestión de residuos sólidos en general (DOCE, 1975). Aunque la ley española era competente sobre los residuos industriales, no regulaba su gestión, ni establecía base alguna para su desarrollo reglamentario. Por lo tanto la gestión de residuos tóxicos y peligrosos (RTPs) no ha existido prácticamente hasta el momento en que se promulgó la Ley 20/1986 de 14 de Mayo, Básica de Residuos Tóxicos y Peligrosos (BOE, 1986), pudiendo afirmarse que antes de esta ley más del 85% de este tipo de residuos eran eliminados incontroladamente.
Estas consideraciones y la circunstancia del ingreso en la CEE, condujeron a la promulgación de dicha ley, que establece la siguiente definición de Residuos Tóxicos y Peligrosos (capítulo 1°, artículo 2º):
“Los materiales sólidos, pastosos, líquidos, así como los gaseosos contenidos en recipientes, que, siendo el resultado de un proceso de producción, transformación, utilización o consumo, su productor destine al abandono y contengan en su composición alguna de las sustancias y materias que figuran en el Anexo de la presente ley en cantidades o concentraciones tales que representen un riesgo para la salud humana, recursos naturales y medio ambiente”.
Quedan excluidas de su ámbito las actividades o los residuos que por sus características poseen un tratamiento legislativo propio, como son los residuos radiactivos, mineros, emisiones a la atmósfera y efluentes cuyo vertido al alcantarillado, a los cursos de agua o al mar esté regulado por la normativa vigente.


Uno de los casos más notable de vertidos no regulados en España son los
que se realizaron en la (“ex”) Bahía de Portman (izquierda). Durante
unos 30 años, hasta fines de los 80’s se vertieron “directamente” los
lodos de rechazo (tailings) de la planta de flotación de la compañía
minera Peñarroya. Debido a esto la bahía se colmató con unos
50 Mt de materiales. A la derecha se puede apreciar el antiguo muelle,
hoy rodeado por tierra.
Esto nos lleva a la problemática de la clasificación de los residuos, base de la legislación y normativa para su manipulación y almacenamiento. En el momento actual, desde el punto de vista legislativo, se establecen los siguientes tipos de residuos según su origen:
– Residuos sólidos urbanos. Son los que se originan en las ciudades y áreas próximas, e incluyen los residuos domiciliarios, los generados en vías urbanas, zonas verdes y recreativas, los de construcción, demoliciones y obras domiciliarias, animales domésticos muertos, muebles y enseres, y vehículos abandonados.

Residuos urbanos.
– Residuos industriales. Los que generan las actividades industriales. Pueden ser muy variados, en función del tipo de industria que los genere, aunque poseen en común la característica de ser potencialmente peligrosos.

Residuos industriales.
– Residuos mineros. Son los que se originan durante las actividades mineras. Incluyen los procedentes de las etapas de prospección, extracción y almacenamiento de recursos minerales, pudiendo ser sólidos o líquidos.

Almacenamiento de residuos sólidos: balsa de estériles (tailings) en Andacollo (Chile).

Almacenamiento de residuos mineros líquidos: estanque de solución
cianurada (flecha amarilla), que descansa sobre un fondo impermeable de
plástico de alta densidad (flecha roja). Punitaqui, Chile.
– Residuos radioactivos. Generados en Centrales Nucleares, y en las instalaciones que utilizan materiales radioactivos, tales como plantas de tratamiento de minerales de uranio, hospitales, etc.


Recubrimiento de las instalaciones de producción y las escombreras de concentrados
de uranio de Andujar (ENUSA).
– Residuos forestales. Son los que proceden o bien del mantenimiento y mejora de las montañas y masas forestales, cuando se hacen podas, limpiezas, etc., o bien de los residuos resultantes de cortar los troncos de los árboles para hacer productos de madera.

Residuos de la industria maderera.
– Residuos agropecuarios. Pueden ser agrícolas o ganaderos, según el tipo de explotación agropecuaria que los origine.


Prensado de aceitunas (izquierda), proceso que deja un residuo sólido contaminante
denominado alpechines. Estos deben ser procesados en plantas de tratamiento
(derecha) antes de su vertido.
– Residuos sanitarios o clínicos. Son los originados en instalaciones sanitarias: hospitales, clínicas, centros de salud, que pueden entrañar extrema peligrosidad.


Residuos hospitalarios.
La mayoría de los residuos antes señalados pueden ser clasificados como potencialmente tóxicos y peligrosos.
Por otra parte tenemos los residuos inertes, que se definen como aquellos que no experimentan transformaciones físicas, químicas o biológicas significativas. No son solubles, ni combustibles, ni reaccionan física ni químicamente de ninguna otra manera, ni son biodegradables, ni afectan negativamente a otras materias con las cuales entran en contacto de forma que pueda dar lugar a contaminación del medio ambiente o perjudicar la salud humana.

Bloques de granito abandonados. No representan ningún peligro en
términos de toxicidad potencial.
La lixiviabilidad total, el contenido de contaminantes, y la ecotoxicidad del lixiviado deberán ser insignificantes en los residuos inertes, y en particular no deberán suponer un riesgo para la calidad de las aguas superficiales y/o subterráneas. Proceden de desechos de obras de reforma, reparación, mantenimiento y nueva construcción de viviendas y otras edificaciones. En ocasiones, en el flujo de residuos de estas actividades también se encuentran sustancias calificables como residuos peligrosos (pinturas, barnices, colas y pegamentos, etc.). El problema que generan estos residuos viene dado por el enorme aumento de su producción y su vertido incontrolado.
Cada uno de estos tipos presenta características propias, y en el momento actual hay un gran interés por ordenar de forma adecuada su eliminación o almacenamiento, lo que se traduce en una importante actividad legislativa que conduzca a una serie de normas para llevar a cabo esta eliminación o almacenamiento. En esta regulación los estudios del terreno y de los materiales que se consideren adecuados para proteger los lugares de almacenamiento es donde la mineralogía ofrece un campo de estudio de gran importancia, para el correcto sellado de los almacenes.
6.2.- El entorno adecuado
El primer paso en la instalación de un almacén de residuos de cualquier tipo es la caracterización de la geología de la zona de interés. Resulta evidente que el suelo y el subsuelo de la zona donde se instale el almacén va a tener que soportar el peso de los materiales que allí se acumulen. Por otra parte tendrá que contribuir en lo posible a evitar que los posibles lixiviados que genere el residuo se dispersen y vayan a parar a los suelos próximos, y a las aguas subterráneas. Por ello, el substrato de la zona debe guardar una serie de características que lo hagan adecuado a este fin. En función de estas características, los posibles emplazamientos pueden ser de tres tipos:
– Impermeables. Aquellos que aseguran un perfecto confinamiento de los residuos y sus lixiviados. Deben estar constituidos por arcillas bentoníticas, margas o rocas compactas no fisuradas, con suficiente espesor como para asegurar la no contaminación de acuíferos profundos.
– Semipermeables. Aseguran una migración lenta de los lixiviados a través de una zona no saturada de espesor suficiente, de manera que permitan una depuración eficaz. Corresponden a formaciones geológicas con porosidad intergranular, por ejemplo, arenosas, y de rocas poco fisuradas.
– Permeables. Permiten una migración rápida de los lixiviados, de manera que no aseguran una depuración eficaz. Corresponden a rocas muy fracturadas, formaciones cársticas, y formaciones de porosidad intergranular con escaso espesor de la zona no saturada.
La Tabla adjunta muestra las características de distintas formaciones geológicas de acuerdo con su permeabilidad, y los tipos de almacenes de residuos que podrían albergar.
| Tipo de emplazamiento | Impermeable | Semipermeable | Permeable |
| Coeficiente de permeabilidad K (m/s) | K < 10-10 en 5 m de espesor | 10-10 < K < 10-6 | K > 10-6 |
| Ejemplos de formaciones geológicas | Arcillas, Margas, pizarras, esquistos | Formaciones areno-arcillosas, areniscas | Gravas, arenas, rocas muy fisuradas y cársticas |
| Valoración | Terrenos favorables, siendo únicamente necesario:
– Drenar los lixiviados – Drenar la escorrentía superficial que llega al almacén |
Terrenos utilizables, siempre que:
– La zona no saturada asegure la depuración eficaz de los lixiviados – Se impermeabilice el vaso de vertido |
Riesgo muy alto de contaminación externa. Terrenos no adecuados |
| Tipos de residuos sólidos admisibles | – Industriales, tóxicos y peligrosos
– Urbanos – Inertes |
– Urbanos
– Asimilables a urbanos – Inertes |
Inertes |
6.3.- El empleo de minerales y rocas en el propio almacén
Una vez establecido que el entorno es adecuado desde el punto de vista geológico para la instalación del almacén, se pasa al estudio de su organización, de forma que se asegure de forma aún más firme que los productos de la lixiviación de los residuos no van a escapar del área controlada. Para ello se diseñan una serie de barreras y capas de seguridad externas e internas, en las que participan determinadas rocas o minerales. Uno de los elementos de seguridad básico en este tipo de almacén es el sellado adecuado de los materiales almacenados. Los dispositivos de sellado que se pueden utilizar en función de las características del residuo almacenado y del substrato sobre el que se dispone son los siguientes:
- Sellado perimétrico. Suele consistir en una barrera que rodea al emplazamiento, completamente o al menos en aquellas zonas que puedan ser susceptibles de escape de lixiviados. Se trata de excavaciones tipo zanja, que se rellenan con una mezcla del propio material que se extrae de la zanja y arcillas, que aseguren una adecuada disminución de la permeabilidad de la berrera. La anchura, profundidad, y calidad de la mezcla de arcillas dependerán de parámetros propios de cada emplazamiento: profundidad prevista del mismo, nivel piezométrico, permeabilidad de la formación en que se implante, distancia al almacén.
- Capa de impermeabilización. Constituye la cubierta inferior del vaso del almacén. Debe incluir una capa mineral que cumpla con los siguientes requisitos de permeabilidad y espesor:
Tipo de residuos Permeabilidad (m/s) Espesor Tóxicos y peligrosos K ≤ 10-9 ≥ 5 m No peligrosos K ≤ 10-9 ≥ 1 m Inertes K ≤ 10-7 ≥ 1 m
Además de esta barrera, deberá existir una sistema de recogida de lixiviados e impermeabilización artificial, que incluya una capa de drenaje de al menos 0.5 m de espesor.



Impermeabilización del fondo de un estanque (izquierda) para impedir las filtraciones en profundidad.
Al centro, bentonitas para el sellado. A la izquierda, aplicación de las bentonitas en el fondo del estanque.
- Recubrimiento superior.Tiene por objeto impedir la dispersión aérea de los residuos almacenados y sus productos (olores, gases), así como impedir el acceso al almacén de las aguas pluviales. En función del tipo de residuo y de la mayor o menor necesidad de asegurar su perfecto aislamiento, deberá tener distintas características de permeabilidad y espesor, siendo los requerimientos más estrictos los correspondientes a los almacenes de residuos tóxicos y peligrosos. Por otra parte, este sellado superior, al impedir la salida de gases, puede suponer un riesgo de acumulación de los mismos, lo que obliga a estas instalaciones a contar con sistemas de extracción (y en su caso, combustión, con o sin aprovechamiento energético) de dichos gases.


Sellado superior de los residuos de una planta de papel (izquierda).
A la derecha un detalle de los materiales sintéticos de sellado.
- Capas de separación.La organización interna del almacén exige la separación física de los sectores en que se dispongan los residuos de cada día, o los de distinta naturaleza. La separación de estos sectores se lleva a cabo con un material impermeabilizante, de unos 15-20 cm de espesor aproximado. Estos materiales representan del orden del 25% del volumen del relleno del vertedero.
- Material compactante.En muchos casos el residuo se almacena conjuntamente con un material que actúa asegurando la compactación del conjunto, y favorece la estanqueidad dentro de los diferentes sectores. Se suelen emplear materiales de procedencias diversas tales como rechazos de canteras o graveras.
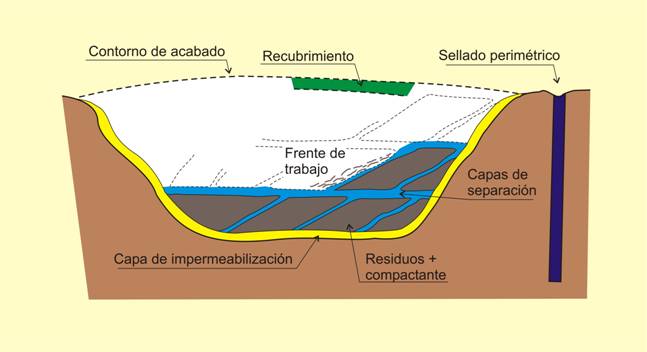
Esquema de un almacén de residuos
En definitiva, este tipo de instalaciones requieren un alto volumen de materiales, cuyas características de permeabilidad deben ser las adecuadas para cumplir las especificaciones establecidas para cada una de las posibles aplicaciones. En general se requieren materiales de baja permeabilidad, lo que por lo general se garantiza con la presencia en el mismo de un alto porcentaje de arcillas.
6.4.- Los almacenes de residuos nucleares: Almacenes geológicos profundos
Los residuos nucleares tienen una problemática propia, por el riesgo que presenta la radioactividad que emiten, y por los largos periodos de tiempo que se requieren para esa radioactividad cese. Por ello, se consideran completamente aparte del resto de tipos de residuos, y para su almacenamiento aún no existe una solución definitiva adecuadamente establecida. Todo esto ha acarreado una amarga polémica entre los diversos estamentos del Estado en países donde existen instalaciones nucleares. Los grupos ambientalistas más radicales no quieren ni siquiera oír hablar de almacenar específicamente los residuos nucleares. Otros sectores de la sociedad están de acuerdo, “pero no cerca de su pueblo”. Aquí es donde se mezclan altas cuotas de hipocresía e ignorancia, mientras las centrales nucleares siguen manteniendo y acumulando sus residuos in situ, sin que por ahora se vislumbre una solución al respecto. En algún momento la sociedad tendrá que decidir si seguir haciendo como el avestruz (escondiendo la cabeza) o enfrentarse al problema de una manera decidida. Mientras tanto prosiguen los estudios técnicos al respecto.
Centrales nucleares en España.
Una de las posibilidades técnicas más ampliamente manejadas es la del enterramiento profundo de estos residuos, en instalaciones adecuadas para:
– Asegurar la estanqueidad del almacenamiento
– Asegurar la seguridad frente a accidentes naturales: fallas, seísmos, impactos meteoríticos.
– Asegurar la seguridad frente a accidentes de carácter humano: guerras, explosiones nucleares.
En este caso, como en el de los residuos no radioactivos, la mineralogía tiene importancia en dos aspectos diferenciados: la selección de la formación geológica adecuada para el emplazamiento del almacén, y el uso de barreras de protección para impedir el escape de los materiales radioactivos almacenados y de sus eventuales productos de lixiviación.
6.4.1.- Formación Geológica albergante
Los requisitos funcionales que debe poseer una formación geológica para servir como almacén de residuos nucleares son los siguientes:
o Asegurar unas condiciones de estabilidad físico-química, hidráulica, mecánica y geoquímica.
o Asegurar un flujo de agua bajo, lento y estable en el repositorio.
o Retardar al máximo o inmovilizar la migración de radionucleidos entre el repositorio y la biosfera.
o Permitir la viabilidad constructiva y operativa del repositorio.
o Asegurar el repositorio frente a la intrusión humana
Para cumplir estos requisitos funcionales las formaciones geológicas albergantes del repositorio deben tener:
o Profundidad y extensión suficiente como para aislar al repositorio de procesos naturales o actividades humanas no deseadas.
o Estabilidad tectónica (carencia de fallas activas) y baja sismicidad.
o Poca complejidad estructural (simplicidad).
o Homogeneidad litológica.
o Baja permeabilidad y gradiente hidráulico. Condiciones adecuadas de retención de radionucleidos (capacidad reductora y de retardo, inmovilización).
o Posibilidad de representar o simular su funcionamiento mediante modelos numéricos.
El abanico de litologías que cumplen estos requisitos es muy amplio, si bien las más extendidas son los emplazamientos graníticos, arcillosos y salinos. Esto sin embargo no excluye otras formaciones geológicas, que en zonas particulares pudieran reunir las condiciones citadas (tobas volcánicas, esquistos, etc.). Las principales características de las litologías citadas son:
o Granitos: Baja permeabilidad, muy baja solubilidad, capacidad de retención variable, resistencia a la alteración química y mecánica, estabilidad tectónica frecuente, conductividad térmica moderada, excavación fácil y estable, elevada resistencia a la erosión y movimiento del agua de tipo advectivo en fracturas (las substancias disueltas se mueven con el agua debido a gradientes hidráulicos).

Granito en el norte de Chile (Chañaral).
o Sales: Muy baja permeabilidad y porosidad, alta conductividad térmica, poca o nula fracturación, naturaleza plástica y propiedades autosellantes, fácil excavación, baja capacidad de retención y movimiento de tipo difusivo (el agua no se mueve pero sí los iones disueltos por gradientes de concentración).


Depósito de sales cloruradas, Cardona, España.
o Arcillas: Muy baja permeabilidad, muy alta capacidad de retención, alta o media plasticidad y capacidad de autosellado, conductividad térmica baja, baja corrosividad, fácil excavación pero con necesidad de estructuras de sostenimiento, poca fracturación y movimiento del agua por difusión.

Arcillas bentoníticas, San José, España.
6.4.2.- Barreras de ingeniería
Para evitar, o retardar lo más posible, que los residuos almacenados se pongan en contacto con el entorno, se emplean una serie de sistemas que actúan como barrera entre la radioactividad y el medio. Para los geólogos es interesante la denominada “barrera de ingeniería de arcilla compactada”, que se coloca entre las cápsulas que contienen los residuos y el medio. Los requisitos funcionales de la barrera de arcilla (bentonita) compactada son los siguientes:
o Minimizar el acceso de agua al resto de las barreras de ingeniería por sellado, al hidratarse, de las fracturas y fisuras generadas en la excavación (retardo en la llegada del agua).
o Estabilizar y homogeneizar la composición química del agua que alcance a los contenedores (constancia en la química del agua).
o Soportar las tensiones mecánicas procedentes de la barrera geológica protegiendo la integridad mecánica de la cápsula (protección mecánica).
o Retardar el transporte de los radionucleidos que puedan liberarse del sistema combustible-cápsula (retardo).
o Disipar adecuadamente el calor de los residuos y el gas generado en la corrosión de los contenedores (disipación de calor).
La arcilla compactada se colocaría rodeando a las cápsulas y en contacto con la formación geológica.

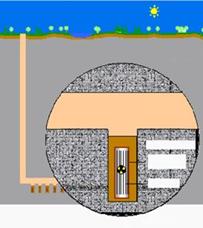
Enterramiento profundo de residuos nucleares en túneles.
A la derecha rellenos con bentonita en los túneles (color marrón).
El material arcilloso seleccionado para fabricar las barreras de arcilla compactada es bentonita. Su selección obedece a:
o Su baja permeabilidad (≈3.10-14 m/s) (minimiza la llegada del agua).
o Su conductividad térmica (0,6-1,4 W/mK) (disipa el calor).
o Su capacidad de hinchamiento (7 Mpa) (sellado de vías de acceso de agua).
o Su superficie específica (725 m2/g) (retención de radionucleidos y conservación de la química del agua dentro de un cierto rango).
o Alta plasticidad (límite líquido 102%-límite plástico 53%) (protección mecánica).
o Capacidad de succión (sellado del repositorio).

Esquema del almacenamiento de residuos nucleares de baja y media actividad
(vida máxima = 300 años) de El Cabril (Córdoba, España): 1) capa filtrante;
2) escollera; 3) arena y grava; 4) arcilla impermeable; 5) cobertura.
6.5.- Bibliografía
Bruhn, D. 2002. Medical wastes at Kandahar dumpsite. http://postconflict.unep. ch/afghanistan/photos/
ENRESA. El almacenamiento geológico profundo de los residuos radiactivos de alta actividad. Principios básicos y tecnología. http://www.enresa.es/Quiosco_ pdf/DDiv_01_ Almacen_geologico_profundo.pdf
Universidad de Navarra. 2004. Centrales nucleares. http://www1.ceit.es/Asignaturas/ Ecologia/Trabajos/enuclear2/nuclear.htm
Waste Magazine On-Line (2004). El Cabril: cementerio nuclear. http://waste.ideal.es/ nuclear.htm
7.- Minerales y Monumentos

Dos de los monumentos más antiguos erigidos por el hombre,
la esfinge y la pirámide de Quefren, mostrando el deterioro progresivo
del pasar de los milenios.
Muchas construcciones humanas están realizadas con piedra natural o poseen componentes minerales: 1) los bloques de las construcciones de mampostería como los chapados de revestimientos interiores y exteriores; 2) objetos decorativos como las estatuas; 3) las lápidas; 4) los ladrillos; y 5) morteros y argamasas están formados en su totalidad o en parte por componentes minerales, que han de soportar la acción de la intemperie, o efectos asociados, por ejemplo, la abrasión, cargas, etc. El mejor uso y la máxima durabilidad de estas construcciones se consigue utilizando en cada caso los materiales más adecuados a cada fin, lo que implica conocer sus propiedades, y prever su evolución futura. Por otra parte, existen un gran número de construcciones antiguas realizadas sin tener en cuenta estos condicionantes, lo que ha producido su degradación.

Esculturas degradadas por el agua y la corrosión química.
En los procesos de degradación se originan componentes minerales que pueden ser muy perjudiciales para la evolución posterior del proceso, por lo que es muy conveniente conocerlos e intentar evitar, en lo posible, su formación y desarrollo en la construcción correspondiente. Así, en el presente tema estudiaremos los monumentos y construcciones arquitectónicas en general desde estos dos puntos de vista: cuales son los procesos de degradación que afectan o pueden afectar a los materiales constructivos bajo la acción de la intemperie, y cual es su comportamiento en cada caso frente a estos procesos.
Organización del capítulo:
7.1.- La Alteración de los monumentos
7.1.1.- Causas del deterioro de las rocas
7.2.- Efectos de la alteración
7.3.- Rocas, minerales y construcción
7.3.1.- Granitos
7.3.2.- Mármoles
7.3.3.- Pizarras
7.3.4.- Areniscas
7.3.5.- Ladrillos
7.3.6.- Morteros
7.4.- Bibliografía
7.1.- La Alteración de los Monumentos
La alteración de los monumentos está causada por un conjunto de procesos que tienen lugar bajo la acción de los agentes meteorológicos, e implican un cambio del aspecto externo, de la forma, o incluso de la estabilidad mecánica de un componente arquitectónico o escultórico. Son procesos variados en el detalle, y tienen lugar bajo diversos condicionantes: la naturaleza de los minerales contenidos en el elemento arquitectónico afectado, la climatología, y el crecimiento de líquenes, entre otros. A estos hay que añadir otro que tiene gran importancia en el ámbito urbano: la agresividad de la atmósfera en contacto con el material en cuestión, que puede llegar a ser muy alta en este ambiente concreto, debido a la presencia de altas concentraciones de gases contaminantes (CO, CO2, SO2), capaces de reaccionar con el agua, formar ácidos y atacar a los minerales. Analizamos a continuación los diversos factores que influyen en este proceso.
7.1.1.- Causas del deterioro de las rocas
Las causas del deterioro de los materiales que forman monumentos pueden ser de dos tipos: externas o internas.
Las principales causas externas serían:
– Agua. Como ya hemos visto en temas precedentes, la acción del agua sobre los minerales puede originar procesos muy diversos, tanto físicos (acción en cuña de ciclos hielo-deshielo), como químicos: disolución e hidrólisis de minerales, fundamentalmente. A su vez, en el caso concreto de los monumentos, su acción se podrá ver potenciada por factores climáticos y de composición del aire, puesto que los componentes de éste pueden pasar al agua. A esto habría que agregarle que los minerales que son disueltos dan origen a otros, los cuales al cristalizar pueden originar presiones en el orden de los MPa (mega pascales). Un caso notable en este sentido es la epsomita, con una presión de cristalización de 290 MPa. No obstante, estas presiones son equivalentes a las que puede ejercer el hielo, de hasta 193 MPa.
– Atmósfera. Los componentes atmosféricos pueden ser muy variados, y pueden en unos casos actuar directamente sobre los minerales (caso del ozono), y en otros aportar agentes que las aguas, o el propio vapor de agua que la atmósfera pueden incorporar, actuando sobre las rocas en forma líquida. Los principales contaminantes atmosféricos son: los óxidos de nitrógeno, carbono y azufre procedentes de la combustión de hidrocarburos, el gas metano emitido por los fertilizantes y la quema de bosques, y los gases de combustión liberados en la incineración de residuos sólidos.
– Organismos vivos (biodeterioro). La acción de los organismos sobre los monumentos puede ser muy variada, e incluye desde fenómenos puramente físicos, como la acción de las raíces de plantas, que pueden introducirse por las grietas o por las juntas de las edificaciones, o afectar a las cimentaciones, o los efectos químicos o físico-químicos producidos por la acción de los excrementos de aves, o por la acción de líquenes o de bacterias.

Deterioro progresivo de la piedra a la intemperie bajo la acción de los elementos y los líquenes.

Edificios invadidos por las raíces de árboles en la ciudad monumental de Angkor Wat (Camboya).
– Antropogénicas. Incluimos aquí los factores relacionados con la acción del hombre, ya sea previamente a la instalación del material, o durante la misma (tipo de labra, tratamientos que reciba, cargas estructurales que se le apliquen, posición geométrica en que se dispongan, tipo de elemento arquitectónico: los suelos sufren efectos muy distintos que las paredes o las fachadas), o tras la misma (instalación de letreros). Por otra parte debemos incluir en este apartado el tráfico. Por un lado, favorece la presencia de contaminantes en la atmósfera, que resultan especialmente agresivos en el ámbito urbano. Por otro, las vibraciones que se relacionan con el tráfico pesado pueden afectar a la estabilidad de las construcciones próximas. A esto debemos agregar las calefacciones que utilizan calderas a carbón, y las refinerías de petróleo que muchas veces están cerca de los núcleos urbanos.


Coches y refinerías de petróleo, dos fuentes de contaminación que se retroalimentan.
– Otros. Como agentes externos de menor importancia en general podemos citar la acción del viento y de las temperaturas (o de sus variaciones extremas).
Por otra parte, tenemos las causas internas, propias de la roca que sufre el proceso o procesos correspondientes:
– Mineralogía. La composición mineralógica del material de construcción es siempre fundamental para explicar las transformaciones pueda sufrir, pues cada mineral presenta distintas susceptibilidades a los agentes externos descritos: unos son fácilmente solubles o hidrolizables, o sufren más la acción de los agentes atmosféricos, o se desgastan con mayor facilidad por presentar menor dureza. Por ejemplo, la calcita, un mineral presente en algunas rocas de construcción, o en el mármol, es fácilmente hidrolizable: CO2 + H2O → H2CO3 y consecuentemente CaCO3 + H2CO3 → Ca2+ + 2 HCO3–. Por su parte, el dióxido de azufre atmosférico produce ácido sulfúrico, que reacciona con la calcita generando yeso.

Muestras de caliza, de grano grueso (izquierda) y fino (derecha).
– Textura. La disposición textural de los minerales puede afectar también a su comportamiento frente a la meteorización. Por ejemplo, las rocas de grano más grueso se degradan, por lo general, más rápidamente que las de grano más fino.


Granito (izquierda) y riolita (derecha). Ambos poseen la misma composición química,
sin embargo el primero posee un tamaño de grano mucho mayor.
– Tectónica. El hecho de que una roca halla sufrido los efectos de deformación tectónica suelen implicar la aparición de diaclasas o microfracturas, que pueden ser prácticamente invisibles. Sin embargo, bajo la acción de cargas se manifiestan por fenómenos de rotura, que a su vez favorecen la acción de otros agentes externos, como la infiltración de soluciones.
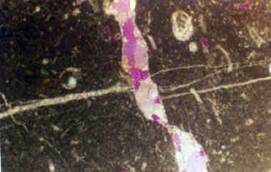
Imagen al microscopio polarizante de una microfractura (centro)
rellena de calcita, en una caliza.
Estas causas actúan siempre combinadas entre sí, produciendo los efectos que veremos a continuación.
7.2.- Efectos de la alteración
Los principales efectos que podemos describir como integrantes del fenómeno definido como “mal de la piedra” se pueden agrupar en tres grandes grupos:
- Efectos que implicanpérdida del material constructivo. Pueden ser de los siguientes tipos:
– Alveolización. Consiste en la aparición de una red bastante continua de cavidades alveolares, que pueden estar originadas por la disolución, o hidrólisis seguida de disolución, de determinados minerales como pueden ser calcita o yeso, o por la acción del viento cargado de partículas, o por la acción de las raíces de plantas. Es relativamente común en calizas, en areniscas con cemento carbonatado, o en rocas porosas en general.

Alveolización de una caliza.
– Excavaciones y cavernas. Consiste en la aparición de huecos individualizados, de ciertas dimensiones (centimétricas como mínimo). Pueden tener el mismo origen que la alveolización, pero suele ocurrir en rocas fundamentalmente no porosas, que sin embargo poseen nódulos de minerales solubles.


Excavación (izquierda) y caverna (derecha) en calizas.
– Erosiones superficiales. Son pérdidas de material rocoso centradas fundamentalmente en la superficie expuesta, que pueden tener las mismas causas que las anteriores, pero suelen ocurrir sobre rocas de carácter masivo.

Erosión (por disgregación) de una arenisca.
– Arenización y disgregación. Se trata de procesos por lo general superficiales, debidos a la pérdida diferencial de algún componente mineralógico, con lo que los demás quedan disponibles para ser liberados. La arenización es característica de los granitos, y se debe a la meteorización de los feldespatos para dar minerales arcillosos, que son eliminados con facilidad, quedando sueltos los granos de cuarzo; la disgregación es típica de las areniscas, por disolución del cemento carbonatado o pérdida de la matriz arcillosa, lo que libera los granos.

Evidencias de arenización en granito.

Disgregación en areniscas.
– Pérdidas de material. En algunos casos llegan a ser generalizadas, perdiéndose completamente determinadas piezas arquitectónicas, normalmente por evolución de alguno de los efectos anteriores.
 Fachada en la que se han perdido bloques completos, que ha habido
Fachada en la que se han perdido bloques completos, que ha habido
que reponer con materiales similares.
- 2.Cambios en la coloración.
– Pátinas. Son delgadas películas superficiales, que pueden tener diversas causas y tipologías:
o Pátinas de envejecimiento: causadas por el propio paso del tiempo y exposición a la intemperie.
o Pátinas de suciedad: ennegrecimiento causado por contaminación ambiental inducida por los hollines residuales dejados por la combustión incompleta de carbones (calefacciones) o del diesel de camiones u autobuses.

Pátina de suciedad en bloques de calizas.
o Pátinas de lavado: producidas por escorrentía diferencial del agua.

Pátina de lavado sobre ladrillería.
o Pátinas de decoloración: varía la tonalidad natural de la piedra, es la llamada «noble pátina».
o Pátinas biogénicas: la superficie de la roca está recubierta por organismos, como pueden ser líquenes.

Pátina biogénica en areniscas.
– Eflorescencias. Son manchas blancas producidas por la precipitación de sales solubles al migrar y evaporarse el agua en la superficie de rocas porosas. La procedencia de las sales es muy diversa; la fuente puede estar en el suelo, en aguas subterráneas, excrementos de aves, antiguos tratamientos, en morteros utilizados o proceder de la roca original, la cercanía al mar, etc. Las sales más comunes en las rocas de los monumentos son los sulfatos, cloruros, carbonatos y nitratos. Si la formación de estas sales tiene lugar bajo la superficie de la piedra reciben el nombre de subeflorescencias, y si se forman en el interior criptoeflorescencias. Su efecto destructor es función de:
o Tipo de sal formada y lugar de cristalización de la misma.
o Condiciones ambientales, la humedad y temperatura controlan los procesos de evaporación, disolución y precipitación.
o Presencia de fracturas o cavidades en la roca, ya que la cristalización de las sales en éstas implica un aumento de volumen que tiende a aumentar el grado de fracturación o del tamaño de las cavidades.

Ejemplo de eflorescencia.
– Costras. Son láminas de material endurecido resultado de la transformación superficial del sustrato pétreo. Estas costras se desarrollan en capas, presentando una morfología, dureza y color determinados y su naturaleza físico-química nada tiene que ver con la del sustrato.
– Depósitos superficiales. Son acumulaciones de material extraño a la roca sobre su superficie. Pueden ser excrementos, o simplemente polvo sobre el que a su vez puede crecer musgo.

Depósito superficial sobre granito.
- Fracturaciones. Pueden ser de diversos tipos:
– Fragmentación. La presencia de facturas no visibles y la acción de cargas estructurales puede hacer que los elementos arquitectónicos queden fragmentados longitudinal o transversalmente.

Caliza con fragmentación acompañada de otros procesos de degradación.
– Exfoliación. Es una fisuración de la roca característica de las rocas que presentan esquistosidad o foliación metamórfica, consistente en la separación a la escala milimétrica y menor, normalmente favorecida por la propia naturaleza de la roca.


Esquisto (izquierda) y pizarra (derecha) mostrando sus planos de exfoliación.
– Separación en placas. Es un fenómeno de exfoliación grosera, normalmente paralela a la superficie de exposición, que puede tener su origen en una meteorización diferencial, de forma que la parte más externa se altera más rápidamente, y llega a separarse físicamente del resto de la roca.

Separación en placas en granito.
7.3.- Rocas, minerales y construcción
Todos hemos visto alguna vez edificaciones o complementos arquitectónicos realizados con materiales naturales (granitos, mármoles, pizarras), afectados por problemas de alteración importantes, que pueden llegar a implicar incluso el riesgo de desmoronamiento. Ello se debe a que estas rocas contenían minerales que han sufrido una meteorización física o química que ha implicado cambios en su comportamiento mecánico, modificando con ello la del conjunto.
Sobre esta base, debemos conocer cuales son las características mineralógicas que afectan a la estabilidad a medio-largo plazo de los minerales que forman las rocas y componentes más empleados en construcción. Esto es de la mayor importancia para protegerlas adecuadamente y así evitar el riesgo de una rápida degradación. Estudiaremos a continuación algunos de los materiales naturales empleados en la construcción para conocer sus problemas específicos: analizaremos las características desde este punto de vista de Granitos, Mármoles, Pizarras, Areniscas, Ladrillos y Morteros.
7.3.1.- Granitos
Los granitos, en un sentido amplio, son rocas de elevada dureza y resistencia física, siempre y cuando no contengan minerales fácilmente alterables que estén siendo afectados por procesos de meteorización. Los principales factores que condicionan las propiedades mecánicas del granito son los siguientes:
– Alterabilidad de los feldespatos. Los feldespatos son los minerales más fácilmente alterables en las rocas graníticas. Cuando están sanos son minerales de alta dureza, muy aptos para un buen pulido, y con muy baja capacidad de absorción de agua. Sin embargo, su alteración genera minerales arcillosos (caolín, illita), que presentan caracteres diametralmente opuestos: baja dureza, no aptos para el pulido, y con muy alta capacidad de absorción de agua. Por ello, la diferencia de comportamiento mecánico, y de durabilidad frente a la intemperie de un granito con feldespatos sanos frente a otros en que los feldespatos están alterados es enorme. Por ejemplo, analicemos la hidrólisis de un feldespato potásico, constituyente normal de los granitos. El primer paso nos llevará a la formación de un mineral tipo mica potásica:
3 KAlSi3O8 + 2 H+ → KAl3Si3O10(OH)2 + 6 SiO2 + 2 K+
pero si hay abundancia de hidrogeniones (medio extraordinariamente ácido), esta fase no será estable, hidrolizándose a su vez, y dando como producto una fase mineral tipo caolín:
2 KAl3Si3O10(OH)2 + 2 H+ + 3 H2O → 3 Al2Si2O5(OH)4 + 2 K+

Granito fuertemente alterado a caolín.
– Alterabilidad de las micas. El granito a menudo contiene micas, siendo la biotita la más típica que la moscovita. Ambos minerales son mucho más blandos que el cuarzo y los feldespatos, y tan alterables como los feldespatos, dando también origen a filosilicatos: clorita por parte de la biotita, illita por parte de la moscovita. Por tanto, la presencia de micas en el granito en grandes cantidades implica un cierto riesgo de que la roca presente problemas de estabilidad mecánica.

Microfotografía de clorita (verde) formándose a partir de biotita.
– Alterabilidad de los anfíboles. Los anfíboles son metaestables en condiciones atmosféricas, y tienden a alterarse con relativa facilidad.
– Alterabilidad de los sulfuros. Las rocas graníticas suelen incluir en su mineralogía sulfuros, normalmente como minerales accesorios. Los más comunes son los de hierro (pirita, marcasita, pirrotina). En determinados casos, y bajo ambientes atmosféricos agresivos estos minerales pueden alterarse, generando sulfatos y óxidos, que dan origen a manchas marrón-rojizas. Esta es una evidencia clara de un proceso de oxidación de los sulfuros, cuyo producto final, después de una secuencia de procesos químicos, es la goethita: 4 FeS2 (pirita) + 14 O2 + 4 H2O → 4 Fe2+ + 8 SO42- + 8 H+, seguido de 4 Fe2+ + O2 + 4 H+ → 4 Fe3+ + 2 H2O, y finalmente Fe3+ + 2 H2O → 4 FeO(OH) (goethita) + 3 H+.

Oxidación incipiente (pátina superficial) de pirita en cubos a goethita.
– Textura. La textura de las rocas graníticas no presenta una influencia clara en la alterabilidad del material, salvo en el sentido general de que las rocas de tamaño de grano más grueso resultan más alterables que las de grano fino.
7.3.2.- Mármoles
Los mármoles están formados mayoritariamente por calcita, acompañada en proporciones variables por dolomita, cuarzo, micas, u otros minerales variados en el detalle. Sin embargo, bajo el punto de vista industrial, reciben el nombre genérico de mármoles un conjunto de rocas que incluyen los mármoles auténticos, calizas, y otras rocas de dureza similar, como la serpentinita, considerada industrialmente como mármol verde.



Distintos mármoles. Los colores rojizos, incluso en venas (derecha) se deben a la presencia de óxidos de hierro.
Desde el punto de vista de su caracterización como roca apta para resistir el entorno medioambiental interesan los siguientes parámetros mineralógicos y afines:
– Calcita. Es un mineral de dureza intermedia-baja, poco resistente a los procesos de meteorización física. También desde el punto de vista químico es un mineral poco estable, que en presencia de agua se hidroliza y disuelve con relativa facilidad. Por tanto, son rocas poco durables en un ambiente externo químicamente agresivo como el de las grandes ciudades.
– Dolomita. Es un mineral algo más resistente que la calcita desde el punto de vista físico y químico. No obstante, la textura de las rocas dolomíticas suele ofrecer tal porosidad que se pierden las ventajas que ofrece la presencia de este mineral.
– Otros minerales. El resto de minerales que podemos encontrar formando parte de un mármol pueden ser muy variados. De entre ellos, los más interesantes pueden ser la brucita(mármoles brucíticos), que tiende a alterarse con relativa facilidad, y los sulfuros de hierro (pirita-pirrotina-marcasita), que como vimos en el caso de las rocas graníticas, pueden causar problemas de formación de manchas por alteración a sulfatos y óxidos. También pueden aparecer venas de óxidos de hierro de color rojizo.
– Textura. La textura de las rocas marmóreas presenta variaciones muy notables, dado que el grupo engloba tanto los mármoles auténticos, caracterizados por texturas granoblásticas, más o menos gruesas, como las calizas, con una enorme variabilidad textural: calizas bioclásticas, oolíticas, micríticas, etc. De estos tipos texturales resultan especialmente problemáticos los que presentan altas porosidades, puesto que permiten y favorecen la entrada de agua, que actúa sobre los carbonatos hidrolizándolos y disolviéndolos.
7.3.3.- Pizarras
Las pizarras son rocas lutíticas afectadas por un metamorfismo de grado bajo. Estas condiciones dan lugar una transformación de la textura que da origen a la esquistosidad, aunque no se produce una blástesis importante de minerales típicamente metamórficos. De esta forma, las pizarras de alta calidad, para techados y suelos nobles, deben estar constituidas casi exclusivamente por micas microcristalinas (illita) y clastos de cuarzo de tamaño arena-limo (cuanto más pequeños mejor).


Tejados construidos con pizarra, a la izquierda, roca sana, a la derecha, pizarras alteradas por los elementos.
Los minerales accesorios que la pizarra puede contener y que representan un riesgo por ser inestables o metaestables en condiciones ambientales son los siguientes:
– Sulfuros. Los sulfuros, en especial los más abundantes: pirita-pirrotina-marcasita, representan, como ya sabemos, un alto riesgo en todas las rocas de construcción por su posibilidad de sufrir hidrólisis y oxidación. Esto es especialmente importante en las pizarras, puesto que durante el proceso se genera acidez, que no solo puede llegar a decolorar la roca, sino que incluso puede llegar a producir otros fenómenos de hidrólisis y descomposición que acaben generando perforaciones: goteras.
– Carbonatos. La presencia de carbonatos en este tipo de roca es altamente perjudicial para su calidad, dado que presentan comportamientos físico-químicos muy distintos a los del resto de componentes de la pizarra, dando origen a alteraciones en el comportamiento general de las piezas que acaban desestabilizándolas y destruyéndolas.
– Textura. La textura de las pizarras no suele presentar grandes variaciones. La presencia de acumulaciones de granos de arena en forma de nivelillos arenosos (la llamada “febra”) suele ser un factor de inestabilidad, pues estos nivelillos presentan una mayor porosidad, y representan puntos de acceso del agua a la roca.
7.3.4.- Areniscas
Las areniscas han sido ampliamente utilizadas en el pasado como elementos arquitectónicos, tanto en sillería como en mampostería. Sin embargo en la actualidad su empleo está muy limitado, debido a que presentan graves problemas de estabilidad (se alteran con gran facilidad). Ello se debe fundamentalmente a que la mayor parte de las areniscas están formadas por dos componentes claramente diferenciados, con comportamientos físico-químicos muy diferentes: el armazón de granos de tamaño arena, formados fundamentalmente por cuarzo, y la matriz o cemento que los empasta, formada por lo general por minerales de la arcilla (en el caso de la matriz) o por calcita (en el caso del cemento). Este armazón, al ser sometido a cargas estructurales, actúa de forma destructiva para el conjunto, pues los granos tienden a clavarse en la matriz o cemento generando tensiones que favorecen a los procesos de disgregación física y química.


Areniscas en un afloramiento (izquierda) y su utilización como elemento
de construcción (derecha).
Los minerales problemáticos en el caso de las areniscas son los siguientes:
– Feldespatos. La presencia de feldespatos entre los clastos (arena-limo) representa un riesgo de transformación en minerales arcillosos que ya conocemos. No suele ser un problema tan grave como en otras rocas, porque por lo general no son abundantes.
– Matriz arcillosa. La matriz arcillosa de las areniscas representa un factor de debilidad muy importante, por el contraste de dureza respecto al armazón de clastos, y porque se disgrega con mucha mayor facilidad que éstos.
– Matriz carbonatada. Representa también un factor de inestabilidad del conjunto de la roca, debido también a su menor dureza, y al hecho de que se hidroliza y disuelve con facilidad.
– Matriz ferruginosa. Suele ser un cemento muy efectivo, pero confiere a la roca un color rojo que no siempre resulta estético. Por otra parte, este tipo de cemento no suele ser tan continuo y generalizado como la matriz arcillosa o el cemento carbonatado, es decir, puede encontrarse en un volumen limitado de materiales, limitando el interés de la explotación canteril.

Matriz ferruginosa (color marrón rojizo) en areniscas.
– Textura. Desde este punto de visita, el principal factor textural de calidad de las rocas areniscas es la granulometría: las areniscas de grano más grueso presentan un gran contraste de tamaño entre clastos y matriz o cemento que resulta muy perjudicial para el conjunto de la roca, favoreciendo la disgregación.

Aspecto textural de una arenisca observado al microscopio de luz polarizada.
Q: granos de cuarzo, C: cemento de calcita.
7.3.5.- Ladrillos
Los ladrillos son piezas cerámicas, obtenidas por la cocción de mezclas dosificadas de materiales entre los que predominan siempre las arcillas. Su estabilidad en el entorno natural depende, además de de los factores externos, de dos factores propios: la formulación empleada para su fabricación, y el proceso seguido para la misma.

Ladrillos, un elemento clásico en la construcción en España.
– En lo que se refiere a la formulación, la materia prima de los ladrillos es una mezcla dosificada de arcillas y arena cuarzosa, que puede llevar otros componentes minerales o bien de forma intencionada (aditivos para dar o quitar color), o porque la materia prima los contenga y no sea fácil eliminarlos. Por ejemplo, en algunos casos la arcilla o la arena contienen carbonatos o materia orgánica, que son componentes indeseados, ya que durante la cocción se comportan de forma distinta a los componentes habituales.

Desconchados por caliche (carbonatos) en un ladrillo.
Efectos de la materia orgánica en los ladrillos. A la izquierda (de arriba hacia abajo): 1) probeta seca (sin cocer); 2) a 850ºC; 3) a 950ºC; 4-6) a 1050ºC. De 4 a 6 se aprecian los efectos de la presencia de materia orgánica, la que no ha podido escapar en forma gaseosa debido al proceso vitrificación de la parte externa, induciéndose así una deformación notable. A la derecha, probetas 4 a 6 mostrando el denominado “corazón negro”, generado por la reducción de Fe3+ a Fe2+ (hematites → magnetita). Parte de una investigación doctoral sobre el tema llevada a cabo por D. Iván Blanco en el Dept. de Cristalografía y Mineralogía de la UCM.
– El proceso de fabricación tiene como paso principal la cocción. Esta se lleva a cabo a temperaturas de entre 600 y 1000ºC, y da origen a la fusión parcial de los componentes de la fórmula, lo que a su vez da origen a un componente cementante que es el que da compacidad al ladrillo. En función de la formulación concreta necesitaremos diferentes temperaturas de cocción, y si ésta no es adecuada obtendremos un producto cuyas propiedades serán distintas a las esperadas.
Antiguo horno para la cocción de ladrillos.
Efectos de la temperatura sobre las características del producto final (de arriba hacia abajo): 1) probeta seca (sin cocer); 2) a 850ºC; 3) a 950ºC; 4) a 1050ºC; 5) a 1150ºC. Debido a la presencia de carbonatos (fundente) y la elevada temperatura alcanzada en el último caso, el ladrillo ha fundido y ennegrecido. Parte de una investigación doctoral sobre el tema llevada a cabo por D. Iván Blanco en el Dept. de Cristalografía y Mineralogía de la UCM.
Sobre esta base, la calidad y durabilidad de los ladrillos dependen de estos dos factores. Los ladrillos cocidos a temperaturas excesivamente bajas para su formulación se desintegrarán con rapidez, desgranándose fácilmente, mientras que los “excesivamente cocidos” serán demasiado rígidos, y por tanto más frágiles frente a las cargas estructurales.
7.3.6.- Morteros
Los morteros son componentes arquitectónicos básicos para dar solidez a las edificaciones uniendo los elementos de construcción (ladrillos, bloques de piedra). Por sus propias características, el mortero tiene que estar en contacto por un lado con los elementos arquitectónicos, y por otro, con la atmósfera, por lo que no deben ser reactivos con los mismos.

Ladrillos unidos por un mortero de arena y cemento. Se puede apreciar el mal acabado
y procesos incipientes de disolución del mortero.
Los morteros están formados por tres componentes básicos: granos (normalmente de arena, o fragmentos de rocas en los más gruesos), un aglomerante (yeso o cemento) y agua, que se dosifican en proporciones variables para obtener una masa plástica que se aplica para su endurecimiento por fraguado del aglomerante. En función de la formulación concreta que se utilice obtendremos un material apto para diferentes posibles aplicaciones: unión de bloques de sillería, enlucido de muros o paredes, rellenos estructurales, etc.
El yeso para morteros se produce de la siguiente manera, primero formando un compuesto llamado hemiedrita deshidratando parcialmente el yeso (mineral):
CaSO4·2H2O + calor → CaSO4·1/2H2O + 3/2 H2O


El producto industrial (hemiedrita; izquierda) que se vende como “yeso”
aunque no se corresponda con la composición química exacta del mineral (derecha).
La hemiedrita es luego mezclada con agua en el lugar de construcción, lo que constituye un proceso de rehidratación espontáneo:
CaSO4·1/2H2O + 3/2 H2O → CaSO4·2H2O + calor
En el caso del cemento la materia prima es el carbonato de calcio, el que se calienta en hornos rotatorios para formar cal:
CaCO3 + calor → CaO + CO2

Horno rotatorio para la transformación de carbonato de calcio en cal.
Al mezclar la cal con agua se forma un hidróxido:
CaO + H2O → Ca(OH)2 + calor
La cal hidratada reacciona paulatinamente con el CO2 atmosférico para dar a su vez carbonato de calcio:
Ca(OH)2 + CO2 → CaCO3 + H2O
Desde el punto de vista de la alterabilidad, los morteros tienen dos posibles problemas: el de la estabilidad de los componentes, y el de la estabilidad del conjunto. En lo que se refiere a los componentes:
– Los granos (arenas o gravas) serán más o menos alterables en función de su naturaleza, como hemos visto con anterioridad.
– El aglomerante presentará su propia problemática en función de su naturaleza: el yeso sufre procesos de alteración por ciclos de hidratación-deshidratación, disgregándose total o parcialmente. Esto es así porque los enlaces entre las moléculas de agua y los grupos sulfato son débiles, por puente de hidrógeno. Tomando en cuenta que el las moléculas de agua se localizan entre capas de sulfato y de iones calcio, es fácil entender que cualquier proceso que implique la adición o substracción de moléculas de agua produzca importantes cambios estructurales en el mineral. El cemento resiste más este tipo de procesos, pero en atmósfera muy agresiva, como la urbana, o bajo la acción de agentes acidificantes, como los excrementos de aves, también se descompone con relativa facilidad. Recordemos la transformación de cal hidratada en carbonato de calcio (en presencia del CO2). Como ya hemos visto en secciones anteriores, el carbonato de calcio es un mineral fácilmente hidrolizable.

Degradación del yeso en muros.
La estabilidad del conjunto del mortero depende por un lado de las características de los componentes, pero también de la formulación concreta, y de la granulometría de los componentes en granos: cuanto mayores sean, mayor facilidad darán a la infiltración de los agentes meteorológicos en los espacios intergranulares, puesto que éstos serán mayores. En lo que se refiere a la formulación, favorecerá la alterabilidad de los componentes mayoritarios en la mezcla en cada caso.
Por otra parte, la degradación del mortero favorece otros procesos:
– Puede dar origen a neoformación de minerales por infiltración de aguas, disolución, y procesos de precipitación.
– La alteración del aglomerante puede implicar cambios de volumen, generando tensiones estructurales.
– La desaparición total del mortero por alteración del aglomerante y desagregación de los granos genera huecos o elimina el recubrimiento de protección para el que se colocó, o elimina el agente de cohesión general del muro, o elemento arquitectónico correspondiente.
– En algunos casos se puede dar una cierta reactividad entre el mortero, o sus productos de alteración, con los elementos que soporta.
7.4.- Bibliografía
Davis, N.: 1980, Where pipes freeze. Alaska Science Forum, Article 443 (Geophysical Institute, University of Alaska Fairbanks), http://www.gi.alaska.edu/ ScienceForum/ASF4/443.html.
La Iglesia, A., González, V., López-Acebedo, V. and C. Viedma, 1996, Salt crystallization in porous construction materials I: estimation of crystallization pressure. Journal of Crystal Growth 177, 111-118.
8.- Minerales, metales, gases y la salud humana y ambiental
Imagen SEM de crisotilo, con las características
fibras “rizadas”.
No existen muchas formas alternativas de mantener lo que “actualmente” llamamos “civilización” en el mundo occidental sin los metales y un sinnúmero de compuestos químicos utilizados en la industria. Como la actitud natural en el ser humano es casi siempre la de solucionar los problemas por eliminación de los mismos, proponemos aquí el siguiente ejercicio mental: pensemos por un instante que sería de nuestra vida cotidiana si eliminásemos la actividad industrial. Entre muchos ejemplos (y sólo señalaremos los relacionados con los denominados metales pesados), los edificios se vendrían abajo (eliminamos el acero), no podríamos tener acceso a la electricidad (eliminamos los cables de cobre), los coches no se podrían fabricar (eliminamos el acero, el aluminio, el cobre), las baterías y pilas serían inviables (eliminamos el plomo, el níquel, el cadmio, el mercurio), los aparatos electrodomésticos no existirían (eliminamos el acero, el cobre), y lo mismo vale para el material científico y médico. Mientras no se desarrollen tecnología alternativas, seguiremos necesitando los metales, incluso aquellos que representan un riesgo real para la salud humana y el medio ambiente.
¿Existen soluciones para los problemas ambientales relacionados con la industria moderna? sí, definitivamente las hay, porque hoy por hoy existen tecnologías que permiten minimizar el riesgo ambiental derivado de la actividad industrial y además existen legislaciones y agencias gubernamentales, que por lo menos en principio, velan porque los potenciales problemas de contaminación no se produzcan o se mitiguen al máximo.

Combustión de carbones para la generación de energía eléctrica.
Intentaremos en este capítulo estudiar el grado de “peligrosidad” de los minerales y metales liberados al medioambiente, y señalaremos algunas de las consecuencias (ambientales, salud humana) que se derivan de las diferentes interacciones. También insistiremos en algunos casos en el tema de especiación, ya que éste es un concepto clave para estimar la peligrosidad real de un determinado contaminante.
Organización del Capítulo:
8.1.- Minerales y metales peligrosos
8.1.1.- El cuarzo y la silicosis
8.1.2.- Asbestos y la asbestosis
8.1.3.- Carbón
8.1.4.- Metales pesados
8.2.- Límites de toxicidad
8.2.1.- Vida acuática en sistemas de agua dulce (ríos, lagos)
8.2.2.- Vida acuática estuarina o en zonas de costas
8.2.3.- Consumo por los seres humanos
8.3.- Efectos ambientales
8.4.- Algunos metales pesados de muy alta toxicidad
8.4.1.- Plomo
8.4.2.- Arsénico
8.4.3.- Mercurio
8.4.4.- Cadmio
8.5.- Gases y aerosoles
8.6.- Bibliografía
8.1.- Minerales y metales peligrosos
Existen dos fuentes principales de introducción de metales al medio ambiente: 1) vía procesos naturales que implican la disolución de la fase mineral que los contiene; y 2) como residuos de la industria minera y otras. Sin embargo, existen minerales que sin contener metales de alta peligrosidad, constituyen en si un riesgo de salud muy importante. Entre estos destacaremos los casos del cuarzo y los asbestos. El primero induce silicosis, y los segundos (asbestos es un nombre genérico) una serie de cuadros clínicos de extrema gravedad. En este caso el medio de propagación es la atmósfera. Las partículas que llegan a la atmósfera constituyen lo que denominamos vulgarmente polvo en suspensión. Su efecto principal es el de oscurecimiento de la misma, pero tiene o puede tener, en función de distintos parámetros, efectos notables sobre la salud de los que lo inhalan. Hay dos cuestiones especialmente relevantes en este sentido: la granulometría de las partículas, y su composición. En lo que se refiere a la granulometría, las partículas de polvo pueden tener tamaños muy variables, en función de la energía que las sustenta. Esta energía puede ser un fuerte viento, o la fuerza de una erupción volcánica, o una voladura de rocas. En cualquier caso, las partículas de tamaños menores se mantienen sistemáticamente durante periodos de tiempo más largos que las mayores. Las más pequeñas tienen mayores “tiempos de residencia” en la atmósfera, aunque todas tienden a sedimentarse en cuanto la energía de sustentación disminuye lo suficiente o cesa. En concreto, las de tamaño inferior a 2.5 μm presentan los mayores tiempos de residencia, con diferencia respecto a las de mayor tamaño. Esto hace que a menudo se estudie la distribución de estas partículas, que pueden tener procedencias remotas. Otra cuestión, que afecta especialmente a la salud, es que las partículas de tamaño inferior a 10 μm son capaces de alcanzar las zonas más profundas del sistema respiratorio (pulmones), mientras que las de tamaño mayor suelen quedar retenidas en el tracto respiratorio. Las menores, por tanto son susceptibles de causar mayores daños orgánicos. Por otra parte, las partículas de estos tamaños menores se suelen originar casi exclusivamente por efecto de procesos de combustión, por lo que suelen ser partículas asociadas a contaminación industrial o urbana. La cuestión composicional tiene también una gran importancia, puesto que algunas partículas pueden producir efectos muy nocivos.
En lo que respecta a los metales, el enfoque de este capítulo estará centrado (aunque no exclusivamente) en las interacciones entre minerales, metales, y los sistemas acuáticos (fluviales, lacustres, estuarinos, y marinos). La razón es simple: 1) el medio acuoso es esencial para que los cationes puedan ser solubilizados; y 2) el medio acuático actúa como un “dispersante natural” de los contaminantes. Así, mientras en un sistema árido los contaminantes tienden a quedar retenidos «in situ», en regiones húmedas, éstos serán rápidamente incorporados a los suelos, para pasar por lavado total o diferencial a las aguas subterráneas, ríos, lagos, o mares, extendiendo así el problema.
8.1.1.- El cuarzo y la silicosis
La silicosis es una de las más antiguas enfermedades laborales, y aun causa la muerte de miles de personas cada año. Se trata de enfermedades pulmonares incurables incluyendo la fibrosis pulmonar y enfisemas, causados por la inhalación de polvo conteniendo sílice cristalina: cuarzo o cristobalita (SiO2).

Polvo de cuarzo sobre un filtro (imagen SEM).
La fuente de esta sílice se encuentra principalmente en las actividades mineras, las que implican la remoción y trituración de rocas en minas, canteras, graveras, o sitios de construcción. Otras actividades que pueden llevar al problema es la limpieza de muros con arena a presión, o la utilización de abrasivos (contengan o no sílice) si se emplean en materiales que contengan sílice. El polvo de sílice cristalina es invisible a simple vista, y es tan ligero que se mantienen en el aire y puede recorrer grandes distancias afectando a poblaciones que en principio no deberían ser de riesgo.
Las medidas de control son relativamente simples, e incluyen el control del polvo que se puede levantar en las actividades mineras por tráfico rodado, voladuras de roca, o perforación, sumemos a esto el uso obligatorio por parte de los operarios mascaras que sean “efectivas” contra el polvo.

Máscara adecuada para la protección contra las partículas de polvo.
Esto último tiene particular relevancia en España, donde el operarios, si no es imposición directa, rara vez emplean los más mínimos sistemas de seguridad.
8.1.2.- Asbestos y la asbestosis
Asbestos el término comercial que se utiliza para una variedad de minerales fibrosos. Dado que las fibras de asbestos son fuertes, de gran duración, e incombustibles, se utilizan ampliamente en la industria, principalmente en la construcción, como materiales aislantes, e ignífugos. La inhalación prolongada de estos materiales por razones laborales puede llevar al desarrollo de: 1) asbestosis (disminución de la capacidad pulmonar); 2) mesotelioma (un tipo de cáncer raro que se desarrolla en la cavidad toráxico o abdominal); o 3) cáncer pulmonar. Existen dos grandes grupos de asbestos, con características, y riesgos de salud diferentes: los asbestos anfibólicos y los asbestos crisotílicos.
Los asbestos anfibólicos (de mayor peligrosidad) contienen más hierro, y resisten los ácidos y altas temperaturas, por lo que se utilizan en hornos industriales y sistema de calefacción. Dos especies comunes se derivan de variedades asbestiformes de anfíboles del tipo:
- Riebeckita (Na2Fe2+Fe3+[Si8O22](OH)2): crocidolita (asbesto azul)
- Cummingtonita ((Mg,Fe2+)7[Si8O22](OH)2) – grunerita
(Fe2+, Mg)7[Si8O22](OH)2): amosita (asbesto marrón).
Por su parte los asbestos crisotílicos se utilizan en materiales de construcción cementantes para techos y como recubrimientos aislantes en cañerías. El crisotilo es un mineral fibroso del grupo de la serpentina (filosilicatos) y su composición química es la siguiente: Mg3[Si2O5](OH)4. Estos asbestos son prácticamente los únicos que se utilizan hoy en día, y la razón hay que buscarla en las estructuras de estos minerales. Los asbestos anfibólicos (crocidolita, amosita) forman fibras tipo varilla, que penetran fácilmente en la pared pulmonar.

Imagen SEM de amosita. Note el carácter acicular de los cristales.
En cambio, las fibras crisotílicas son rizadas, y por lo tanto pueden ser expulsadas con mayor facilidad.

Imagen SEM de crisotilo. Note el carácter “rizado” de las fibras.
Esto, en cualquier caso, no significa que la exposición prolongada a fibras crisotílicas no conlleve peligro, sino que éste será menor en comparación con las fibras anfibólicas.
8.1.3.- Carbón
Aunque el carbón no es una especie mineral en sí, es conveniente por razones prácticas tratarlo como tema de salud ambiental en esta sección. El carbón de consumo doméstico e industrial tiene profundas y peligrosas consecuencias para la salud humana y ambiental. Primero está su uso en las plantas térmicas para producción de energía eléctrica. Muchos carbones poseen contenidos importantes en pirita, la cual en el proceso de combustión genera gases del tipo SO2-SO3. El problema radica en que estos gases reaccionan con las gotas de agua en la atmósfera formando ácido sulfúrico, causante a su vez de la llamada lluvia ácida, con nefastas consecuencia para la biota:
SO2 + H2O = H+ + HSO3–
2 HSO3– = 2 H+ + 2 SO42-

Efectos de la lluvia ácida sobre un bosque.
Otro aspecto negativo del uso de carbones, y causante de muchas muertes en los llamados países del tercer mundo, es la generación de monóxido de carbono, durante la combustión de los residuos. Si la habitación que está siendo calentada por un hornillo de carbón no tiene condiciones mínimas de aireación, las consecuencias pueden ser fatales. Finalmente, un aspecto menos conocido de los carbones son sus altas concentraciones en metales pesados. Por ejemplo, por lo menos 3000 personas en la provincia de Guizhou en China sufren de envenenamiento severo por arsénico. La principal fuente del arsénico parece ser el consumo de guindillas (chiles) que son secados mediante fuegos inducidos por carbones ricos en arsénico. Dichos carbones pueden contener hasta 35.000 ppm de As.
8.1.4.- Metales pesados
Se habla mucho de los metales pesados, sin indicarse sin embargo, qué son, y específicamente, el cómo y por qué son peligrosos. Se denomina metales pesados a aquellos elementos químicos que poseen un peso atómico comprendido entre 63.55 (Cu) y 200.59 (Hg), y que presentan un peso específico superior a 4 (g cm-3). Cabe destacar que en esta categoría entran prácticamente todos los elementos metálicos de interés económico, por tanto, de interés minero.
Lo que hace tóxicos a los metales pesados no son en general sus características esenciales, sino las concentraciones en las que pueden presentarse, y casi más importante aun, el tipo de especie que forman en un determinado medio. Cabe recordar que de hecho los seres vivos “necesitan” (en pequeñas concentraciones) a muchos de éstos elementos para funcionar adecuadamente. Ejemplos de metales requeridos por el organismo incluyen el cobalto, cobre, hierro, hierro, manganeso, molibdeno, vanadio, estroncio, y zinc. El caso del hierro es notable entre éstos, siendo vital para la formación de hemoglobina.
Todos los metales pesados se encuentran presentes en los medios acuáticos (el agua químicamente pura no existe en la naturaleza), aunque sus concentraciones (en ausencia de contaminación) son muy bajas. Los metales pesados se encuentran en estas aguas como coloides, partículas minerales (sólidos en suspensión), o fases disueltas (cationes o iones complejos). Las formas coloidales suelen dar lugar a la formación de hidróxidos, mientras que las partículas sólidas incluyen una gran variedad de minerales. Las fases disueltas pueden a su vez ser capturadas por adsorción o absorción en arcillas o hidróxidos. Adicionalmente, los compuestos orgánicos pueden constituir fases con gran capacidad de captura de cationes metálicos, que en ocasiones dan lugar a fases extremadamente tóxicas (e.g. metilmercurio: CH3Hg).
A su vez la química del sistema acuoso regula las tasas de adsorción-absorción en el sistema agua-sedimento. La adsorción remueve el metal de la columna de agua; la desorción lo incorpora nuevamente a ésta. Los parámetros que regulan el sistema son: la salinidad, el potencial redox (Eh), y el pH:
- Un incremento de la salinidad conlleva una competencia, entre metales pesados y metales grupos I y II, por los sitios de ligazón (e.g. espaciado interlaminar en las arcillas), lo que se traduce en la expulsión de los metales pesados, y su devolución a la columna de agua.
- Un incremento del Eh genera la inestabilidad de los compuestos reducidos (e.g. sulfuros), poniendo el metal en solución.
- Un decrecimiento del pH tiene dos efectos: 1) induce la disolución de compuestos metal-carbonato (e.g. cerusita: PbCO3); y 2) aumenta la solubilidad de los metales disueltos.
El decrecimiento del pH puede ligarse directamente a la serie de fenómenos físico-químicos que se derivan de la oxidación de especies sulfuradas (particularmente la pirita: FeS2). La consecuencia directa es la formación del denominado drenaje ácido. El sistema se encuentra así fuertemente regulado por: 1) las cantidades iniciales de pirita en el yacimiento (de sulfuros o carbones piritosos) o la escombrera (mineral dump); 2) por la presencia de bacterias oxidantes (e.g. T. ferrooxidans); y 3) los niveles de oxígeno.
8.2.- Límites de toxicidad
La Agencia de Protección Ambiental (EPA) de los Estados Unidos ha determinado una serie de límites para las concentraciones de metales pesados. Por encima de éstos los metales pueden causar graves trastornos en los seres vivos, y finalmente ocasionar la muerte. A continuación mostraremos dichos límites en distintos medios y las dosis máximas para la ingesta en los humanos.
8.2.1.- Vida acuática en sistemas de agua dulce (ríos, lagos):
| Metal | Dureza del agua (mg/l) | Límite máximo (μg/l) |
| As | 50 | |
| Be | 130 (+) | |
| Cd | 50 | 0.66 (*) |
| 150 | 1.10 (*) | |
| 200 | 2.00 (*) | |
| Cu | 50 | 6.50 (*) |
| 150 | 12.00 (*) | |
| 200 | 21.00 (*) | |
| Hg | 0.012 (*) | |
| Ni | 50 | 56.00 (x) |
| 150 | 96.00 (x) | |
| 200 | 160.00 (x) | |
| Pb | 50 | 1.30 (*) |
| 150 | 3.20 (*) | |
| 200 | 7.70 (*) | |
| Zn | 50 | 180.00 (#) |
| 150 | 320.00 (#) | |
| 200 | 570.00 (#) |
+: concentración promedio por 1 hora; x: concentración promedio en 24 horas;
*: concentración promedio en 4 días; #: niveles que no pueden excederse en
ningún lapso de tiempo.
8.2.2.- Vida acuática estuarina o en zonas de costas:
| Metal | Límite máximo (μg/l) |
| As | 50 |
| Cd | 8 (*) |
| Cu | 2.9 (+) |
| Hg | 0.025 (*) |
| Ni | 7.10 (x) |
| Pb | 5.8 (*) |
| Zn | 76.6 (*) |
+: concentración promedio por 1 hora; x: concentración
promedio en 24 horas; *: concentración promedio en 4 días.
8.2.3.- Consumo por los seres humanos:
| As | 0.05 mg/l (+) |
| Cd | 10 μg/l (*) |
| Cr | 0.05 mg/l (+) |
| Cu | 1.0 μg/l (#) |
| Hg | 144 ng/l (*) |
| Ni | 632.0 μg/l (*) |
| Pb | 50.0 μg/l (*) (adultos) |
| Zn | 5.0 μg/l (*) |
*: criterios para el agua; +: máximo nivel de
contaminación; #: nivel que jamás debe ser superado.
8.3.- Efectos ambientales
Los organismos pueden verse severamente afectados por pequeñas concentraciones de elementos pesados (ver punto 2). En el caso de los organismos acuáticos, puede que unos determinados valores no induzcan su muerte, sin embargo desarrollarán una serie de problemas fisiológicos y metabólicos (a estas dosis se les denomina subletales). Entre estos problemas podemos mencionar:
- Cambios histológicos o morfológicos en los tejidos.
- Cambios en la fisiología como supresión del crecimiento y desarrollo, torpeza para nadar, etc.
- Cambios en la bioquímica del organismo, tales como en la actividad enzimática, y química de las sangre.
- Trastornos del comportamiento.
- Cambios en la reproducción.
Algunos organismos pueden regular las concentraciones de metales presentes en sus tejidos. Por ejemplo, los peces y crustáceos pueden excretar metales esenciales para su metabolismo (e.g. Cu, Zn, Fe), siempre y cuando éstos superen las dosis requeridas. Desgraciadamente otros metales (no esenciales) tales como el mercurio o el cadmio son excretados con mayor dificultad. Las plantas acuáticas (algas) y los bivalvos (e.g. mejillones, ostras) no son capaces de regular con éxito las concentraciones de metales pesados, y de ahí puede derivarse una serie de problemas. Así por ejemplo, el mercurio puede hacer decrecer dramáticamente la capacidad de fotosíntesis de un alga (e.g. Macrocystes). Los bivalvos por su parte acumulan los metales pesados, pudiendo pasar éstos directamente al ser humano por ingesta. De ahí que se deban tomar precauciones extremas para el consumo en zonas sujetas altos niveles de contaminación (zona de vertidos industriales, metalúrgicos, mineros).
Las vías de incorporación de los metales pesados a los organismos acuáticos son las siguientes:
- Cationes metálicos libres que son absorbidos a través de los órganos respiratorios externos (agallas), los cuales pasan directamente a la sangre.
- Cationes metálicos libres que son adsorbidos por el cuerpo y luego pasivamente difundidos al torrente sanguíneo.
- Metales que son adquiridos durante la ingesta de organismos (otros peces, bivalvos, o algas) contaminados.
En el caso de las algas, el proceso ocurre por absorción a través de las paredes celulares y difusión posterior.
8.4.- Algunos metales pesados de muy alta toxicidad
A continuación nos centraremos en el estudio de los problemas ambientales y de salud humana relacionados con cuatros casos concretos: plomo, arsénico, mercurio, y cadmio. Cabe destacar que no analizaremos todas las fuentes de contaminación, sino solamente aquellas relacionadas con la actividad minera.
8.4.1.- Plomo
El plomo se encuentra presente en un gran número de minerales, siendo la forma más común el sulfuro de plomo (galena: PbS). También son comunes, aunque en orden decreciente, la cerusita (PbCO3) y la anglesita (PbSO4).


Galena (izquierda) y cerusita (derecha).
El plomo es un metal difícilmente movilizable, y bajo condiciones oxidantes la galena da origen a minerales tales como la cerusita y anglesita:
PbS + CO2 + H2O + 2 O2 → PbCO3 + SO4-2 + 2 H+
2 PbS + 4 Fe3+ +3 O2 + 2 H2O → 2 PbSO4 + 4 Fe2+ + 4 H+
Así, el principal riesgo relacionado con la minería del plomo no radica en la posible puesta en solución de este metal (precipita rápidamente como carbonato o sulfato), sino en lo que concierne a los procesos metalúrgicos de las menas de plomo (fundiciones). Cabe destacar que el problema con el plomo no es nuevo (ni siquiera de comienzos de la revolución industrial). Estudios en Suecia revelan que por lo menos el 50 % de la contaminación en suelos del país fue depositada en períodos anteriores al año 1800. El particulado de plomo relacionado con problemas metalúrgicos constituye el problema principal, pero existen otras fuentes que entrañan también una peligrosidad extrema. En los años 90 se constató en la ciudad de Antofagasta (Chile) que había niños que presentaban altos contenidos de plomo en sangre.

Imagen del puerto de Antofagasta (Chile).
La fuente del problema pudo ser determinada, y eran minerales y concentrados de plomo que se acumulaban sin protección en las instalaciones portuarias (pertenecientes a Bolivia), para su posterior envío. Esto nos lleva a encaminar nuestra mirada no solo a las fundiciones, sino también a las zonas donde se acumulan minerales o concentrados de plomo.
El particulado fino de plomo (10-100 μm) puede ser extremadamente peligroso por las siguientes razones:
- Se adhiere más fuertemente a la piel.
- Es más soluble que el particulado grueso en el tracto gastrointestinal.
- Es fácilmente absorbible a través del sistema respiratorio.
El plomo es un metal carente de valor biológico, es decir, no es requerido para el funcionamiento normal de los seres vivos. Debido a su tamaño y carga, el plomo puede substituir al calcio (Pb2+: 0.84 Å; Ca2+: 0.99 Å), y además de manera preferente, siendo su sitio de acumulación, los tejidos óseos. Esta situación es particularmente alarmante en los niños, que debido a su crecimiento incorporan altas cantidades de calcio. Altas dosis de calcio hacen que el plomo sea «removido» de los tejidos óseos, y que pase a incorporarse al torrente sanguíneo. Una vez ahí puede inducir nefrotoxicidad, neurotoxicidad, e hipertensión. Niveles de plomo en sangre de 0.48 μg/l pueden inducir en los niños:
- Daño durante el desarrollo de los órganos del feto.
- Daño en el sistema nervioso central.
- Reducción de las habilidades mentales e iniciación de desordenes del comportamiento.
- Daño en las funciones del calcio (anteriormente mencionado).
- A su vez, niveles del orden de 1.2 μg/l pueden inducir:
- Descenso del coeficiente intelectual (CI). Problemas de desarrollo cognitivo y del comportamiento.
- Déficit neurológicos que pueden persistir hasta la adolescencia.
- Elevación de los umbrales auditivos.
- Peso reducido en recién nacidos. Desarrollo cognitivo temprano anormal.
En adultos que trabajan en ambientes expuestos a la contaminación con plomo, el metal puede acumularse en los huesos, donde su vida media es superior a los 20 años. La osteoporosis, embarazo, o enfermedades crónicas pueden hacer que éste plomo se incorpore más rápidamente a la sangre. Los problemas relacionados con la sobreexposición al plomo en adultos incluyen:
- Daño en los riñones.
- Daño en el tracto gastrointestinal.
- Daño en el sistema reproductor.
- Daño en los órganos productores de sangre.
- Daños neurológicos.
- Abortos.
8.4.2.- Arsénico
El arsénico se encuentra presente en más de 200 especies minerales, siendo la arsenopirita (FeAsS), la enargita (Cu3AsS4), y la tennantita (Cu12As4S13) las más comunes.


Arsenopirita (izquierda) y enargita (derecha).
Por razones no determinadas, la arsenopirita es muy común en los yacimientos minerales europeos (e.g. sulfuros masivos de la faja pirítica de España-Portugal), mientras que la enargita lo es en los yacimientos de la cadena andina, donde constituye una mena principal de cobre (pórfidos cupríferos y epitermales de Au-Ag). La solubilización de las formas sulfuradas de arsénico no es fácil. Esto es claro en el caso de la arsenopirita, la que por ser en ocasiones portadora de inclusiones de oro, ha constituido un tema de numerosos estudios con resultados poco claros hasta la fecha. La reacción fundamental en medio ácido es:
4 FeAsS + 13 O2 + 6 H2O → 4 H3AsO4 + 4 FeSO4
Si además hay pirita en la mena, entonces el sulfato férrico producido actuará de la siguiente manera, coayudando a la oxidación-lixiviación del arsénico:
2 FeAsS + Fe2(SO4)3 → 2 H3AsO4 + 4 FeSO4 + H2SO4
El arsénico puede precipitar finalmente como FeAsO4.
Sin restarle importancia al problema de la solubilización de especies minerales arsenicales, la principal fuente de contaminación está relacionada, al igual que en el caso de plomo, con el tratamiento metalúrgico de los minerales de arsénico. En concreto, los procesos de fundición de concentrados de cobre, que incluyan la presencia de minerales arsenicales (e.g. enargita), pueden dar lugar a intensos problemas de contaminación por vía aérea (arsénico que escapa por las chimeneas), en la forma de As2O3.

Labores mineras (A) y el tostador de minerales de arsénico (B) en El Indio (Chile).
El arsénico que así escapa se deposita luego en los suelos del entorno de la fundición. Dependiendo del volumen de las emisiones y el régimen de vientos, el problema puede extenderse por decenas de kilómetros y más. Un caso notable en este sentido eran por ejemplo las emisiones de la fundición de Chuquicamata (Chile; CODELCO) (parte de los minerales de cobre tratados son arsenicales), con valores de 2340 toneladas/año en 1994. En la actualidad CODELCO (en todas sus divisiones) tiene que recuperar al menos una parte importante del arsénico que potencialmente sería emitido. En Chuquicamata el proceso se realiza en una planta hidrometalúrgica que recupera el cobre, y precipita el arsénico como arsenato férrico. A partir del 2003 las cifras de emisión no podrán exceder las 400 toneladas/año.
El arsénico en los suelos puede ser disuelto y adsorbido por los oxihidróxidos de de hierro (e.g. goethita), las arcillas, o la materia orgánica. Muchos de estos procesos son mediados por la materia orgánica la que puede producir transformaciones del tipo:
- Cambios de redox que inducen la transformación arsenito-arsenato.
- La reducción y metilación del arsénico.
- La biosíntesis de compuestos de arsénico.
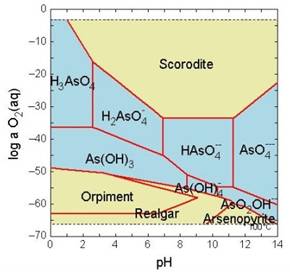
Especiación (minerales y aniones complejos) del arsénico en función del Eh y pH.
La captación de arsénico por parte de minerales tipo goethita proviene de las propiedades coloidales de esta última. Dado que la goethita genera un coloide positivo, éste puede adsorber aniones complejos, que es la forma en que se presentan las especies acuosas de arsénico (ver figura superior).

Imagen SEM-EDX de goethita (Río Toro, Chile) con arsénico adsorbido (sectores brillantes).
La goethita es estable en condiciones oxidantes. Si estas cambian a reductoras, y existe azufre disponible, la goethita se “piritizará” (paso de FeO(OH) a FeS2), y los oxianiones de arsénico (ver figura especiación de arsénico) quedarán libres en solución nuevamente. Un fenómeno químico-mineralógico de esta naturaleza parece ser la causa de la gravísima contaminación por arsénico de las aguas subterráneas en Bangladesh.
Zonas de aguas subterráneas contaminadas con arsénico en Bangladesh.
Las formas solubles metiladas del arsénico (e.g. ácidos metil arsónico [MMA] y dimetil arsínico [DMA]) son fuertemente tóxicas. La ingestión de grandes dosis lleva a problemas gastrointestinales, cardiovasculares, disfunciones del aparato nervioso, y finalmente a la muerte. Recordemos que el arsénico ha sido uno de los venenos de largo plazo más utilizados en la historia de la humanidad, siendo Napoleón (el emperador de Francia), la víctima más famosa. Dosis bajas pero sostenidas (e.g. causas laborales) superiores a 0.75 mg m-3 por año (e.g. 15 años con concentraciones de 50 μg m-3) pueden llevar al desarrollo de cánceres. Resumiendo, entre los principales problemas de salud humana podemos mencionar:
- Problemas gastrointestinales
- Cardiovasculares
- Disfunciones en el sistema nervioso
- Efectos cancerogénicos
- Queratosis
 Queratosis inducida por ingesta sostenida de arsénico.
Queratosis inducida por ingesta sostenida de arsénico.
La vida acuática y terrestre muestra una amplia gama de sensibilidades a las distintas especies arsenicales. En general las formas inorgánicas son más tóxicas que las orgánicas, y el arsenito más peligroso que el arsenato. Los arsenitos pueden fijarse a las proteínas, mientras que el arsenato afecta a la fosforilización oxiditaviva (en relación con Ciclo de Krebs). Los organismos marinos contienen residuos arseniacales que van desde < 1 a 100 mg k-1, los cuales se encuentran como arsenoazúcares (en las algas) o arsenobetaina (en invertebrados y peces). Las plantas terrestres pueden acumular arsénico por captación a través de las raíces, o por adsorción de arsénico aerotransportado, en las hojas.
8.4.3.- Mercurio
La forma principal de mercurio en la naturaleza es el cinabrio (HgS), el que constituye la mena principal para la obtención de este metal. Otras formas minerales incluyen la corderoita (Hg3S2Cl2), la livingstonita (HgSb4S8), y formas supergénicas tales como el mercurio nativo (Hg0), el calomelano (HgCl2), y la schuetteita (Hg3(SO4)O2).


Cinabrio (izquierda) y schuetteita (mineral amarillo; derecha).
El distrito minero de Almadén en España, el más importante del mundo en términos históricos y de producción, posee una mineralogía muy simple que incluye cinabrio como mena mercurial.
Geología del distrito minero de Almadén (España).
El único mineral supergénico de mercurio reconocido en el distrito es la schuetteita, la que aparece como costras recubriendo rocas en las proximidades a escombreras de mineral (mineral dumps).
El mercurio posee una de las peores reputaciones entre los metales pesados. El incidente de la Bahía de Minamata (Japón, años 50s-60s) bastó para que este elemento infundiese alarma pública en todas las regiones del mundo donde podía haber fuentes de contaminación. Consideraciones económicas aparte, todas las investigaciones indican claramente que el mercurio puede constituir una amenaza para la salud humana y la vida silvestre. El riesgo viene determinado por los siguientes factores:
- El tipo de exposición al mercurio.
- La especie de mercurio presente, ya que algunas son más tóxicas que otras, por ejemplo, las formas metiladas de mercurio.
- Los factores geoquímicos y ecológicos que influencian la forma de migración del mercurio en el medioambiente, y los cambios que puede sufrir durante dicha migración.
El cinabrio, aunque es una forma relativamente estable de mercurio, puede también sufrir transformaciones que resultan en especiaciones indeseables. Así, en medio ácido y oxidante tenemos:
HgS → S0 + Hg2+ + 2e–
Esta reacción pone en solución al mercurio, el que puede así puede formar complejos con la materia orgánica (peligrosidad). No obstante, en un medio alcalino oxidante el mercurio precipitará como óxido:
Hg + 2 OH– → HgO + H2O + 2e–
lo que en principio parece una forma más o menos estable, mientras el sistema mantenga la alcalinidad y condiciones oxidantes.
La principal fuente de contaminación con mercurio, en relación con la actividad minera, viene de los gases emitidos por las plantas de tratamiento de cinabrio. El mercurio gaseoso emitido por los hornos (especialmente en los antiguos procesos de tratamiento), es depositado en los suelos que rodean a las instalaciones metalúrgicas como Hg2+. Esto puede ocurrir por depositación directa de emisión de Hg2+ o por conversión de vapores de Hg0 a Hg2+, proceso este último mediado por el ozono (g: fase gaseosa; aq: fase acuosa; p: fase particulada):
Hg0 (g) → Hg0(aq)
Hg0(aq) + O3(aq) → Hg2+(aq)
Hg2+(aq) + hollín/posible evaporación → Hg2+(p)
La reacción fotolítica de Hg2+ a Hg0 en la superficie del suelo puede a su vez contribuir de manera significativa a la emisión de mercurio gaseoso a la atmósfera. Por otra parte, aun cuando el mercurio en el suelo se ligue a una matriz orgánica (ácidos fúlvicos y/o húmicos), el elemento se verá sujeto a procesos de fotoreducción, lo cual también contribuirá a la entrega de mercurio gaseoso a la atmósfera.
De todas las especies de mercurio conocidas, la más peligrosa es sin duda el metilmercurio (CH3Hg). Aunque la forma exacta en que se produce la metilación del mercurio se desconoce, se sabe que en el proceso intervienen bacterias que participan en el ciclo SO42- – S2-. Estas bacterias, que por lo tanto contendrán metilmercurio, son consumidas por el peldaño superior de la cadena trófica, o bien lo excretarán. En este último caso el metilmercurio puede ser rápidamente adsorbido por el fitoplancton y de ahí pasar a los organismos superiores. Debido a que los animales acumulan metilmercurio más rápido de lo que pueden excretarlo, se produce un incremento sostenido de las concentraciones en la cadena trófica (biomagnificación). Así, aunque las concentraciones iniciales de metilmercurio en el agua sean bajas o muy bajas, los procesos biomagnificadores acaban por convertir el metilmercurio en una amenaza real para salud humana.
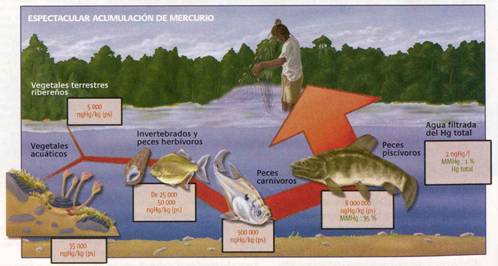
Biomagnificación de las concentraciones de mercurio en el medio acuático.
El metilmercurio daña al organismo de las siguientes maneras:
- Afecta al sistema inmunológico
- Altera los sistemas genéticos y enzimáticos
- Daña el sistema nervioso: coordinación, sentidos del tacto, gusto, y visión.
- Induce un desarrollo anormal de los embriones (efectos teratogénicos); los embriones son 5 a 10 veces más sensibles a los efectos del mercurio que un ser adulto.
En este momento es además tema de debate si otro compuesto mercurial, el thimerosal (C9H9HgNaO2S, un aditivo preservante en muchas vacunas) puede inducir a cuadros de autismo en los niños.
8.4.4.- Cadmio
Los minerales de cadmio, no se encuentran en concentraciones y cantidades suficientes como para justificar una actividad minera específica por el elemento. Entre los minerales de cadmio, la greenockita (CdS) es el más común. Este mineral se encuentra casi siempre asociado con la esfalerita (ZnS). De esta manera, el cadmio se recupera principalmente como un subproducto de la minería, fundición, y refinación del zinc, y en menor grado de la del plomo y cobre. En promedio se recuperan unos 3 kg de cadmio por tonelada de zinc.
Debido a su toxicidad, el cadmio se encuentra sujeto a una de las legislaciones más severas en términos ambientales y de salud humana. En la vida acuática, el cadmio puede incorporarse a los peces a través de dos rutas principales:
- Ingestión
- Introducción en las agallas.
El cadmio así adquirido se acumula en el hígado, riñones, y en el tracto gastrointestinal, y sus efectos son los siguientes:
- Problemas en las agallas y riñones.
- Pobre mineralización de los huesos.
- Anemia.
- Crecimiento retardado.
- Anormalidades del desarrollo y comportamiento.
En el caso de los humanos, el cadmio se puede adquirir por dos vías: ingestión e inhalación. Sus efectos pueden ser divididos en dos categorías:
- Agudos: fiebre de vapores de metal (metal fume fever) causada por una exposición severa; los síntomas son equivalentes a los de la gripe; en 24 horas se desarrolla generalmente un edema pulmonar agudo, el que alcanza su máximo en 3 días; si no sobreviene la muerte por asfixia, el problema puede resolverse en una semana.
- Crónicos: la consecuencia más seria del envenenamiento por cadmio es el cáncer. Los efectos crónicos que primero se observan son daño en los riñones. Se piensa que el cadmio es también el causante de enfisemas pulmonares y enfermedades de los huesos (osteomalcia y osteoporosis). Los problemas óseos han sido observados en Japón (recordar también el problema con metilmercurio; Incidente Minamata), donde se les denominó como la enfermedad itai-itai (por consumo de arroz contaminado con cadmio; causa: irrigación). Otros problemas incluyen anemia, decoloración de los dientes, y pérdida del sentido del olfato (anosmia).
8.5.- Gases y aerosoles
La dispersión de gases distintos a los habituales en la atmósfera es un factor muy importante a considerar. Esos gases contaminantes pueden ser de dos tipos: gases contaminantes “primarios”, es decir, que se emiten de esa forma a la atmósfera, y gases contaminantes “secundarios”, que se forman a partir de reacciones que implican a los primarios. Los más importantes, sus orígenes y efectos son los siguientes.
CO2: un gas común en la atmósfera, pero su excesiva abundancia puede ser letal, por bloquear las funciones respiratorias, induciendo la muerte por asfixia. Esto último puede producirse localmente por acumulación de CO2 en lugares cerrados, sobre todo si hay algún foco local: combustión. También la actividad volcánica suele producir la emisión de enormes volúmenes de este gas, en forma de emanaciones que pueden llegar a ser letales (caso del Lago Nyos, en Camerún). Otro efecto importante de este gas es el temido “efecto invernadero”, debido a su acumulación en la atmósfera a gran escala, produciendo un “oscurecimiento” de la capa atmosférica que permite la entrada de la radiación solar, pero no la salida del calor emitido por el terreno como consecuencia de esta irradiación. Con respecto a su origen antropogénico, se emite como consecuencia todos los procesos que implican combustión: a gran escala, en la obtención de energía eléctrica a partir de combustibles fósiles, y en los vehículos de transporte. En el ámbito minero, afecta tanto a las explotaciones de carbón, como a la utilización de maquinaria pesada con gran consumo de combustibles derivados del petróleo, principalmente diesel en este caso.
SOx, reciben esta denominación genérica los distintos compuestos que resultan de la combustión de compuestos sulfurados: SO, SO2, SO3. Son muy comunes como producto de la combustión de combustibles fósiles, y de la metalurgia de sulfuros por calcinación (pirometalurgia). Su principal problema es que reaccionan con el agua, incluso con el vapor de agua atmosférico, para dar origen a ácido sulfúrico, en áreas con gran actividad industrial o con tráfico muy denso. La lluvia ácida es el más espectacular de los fenómenos asociados a este proceso. La otra fuente importante de óxidos de azufre es la actividad volcánica. En concreto, la lluvia ácida es una consecuencia de la reacción entre este tipo de contaminantes y el agua de lluvia, a través de las siguientes reacciones, formadoras de aniones ácidos:
SO2(g) + H2O(l) = SO2(l)
SO2(l) + 2 H2O(l) = H3O+ + HSO3–
HSO3– + H2O(l) = H3O+ + SO32-
NOx, al igual que los anteriores, se trata de distintos compuestos de nitrógeno originados durante procesos de combustión. Su presencia en la atmósfera representa un problema porque favorece la formación de otros importantes contaminantes, como el ozono o los aldehídos.
El ozono (O3) es un contaminante secundario que se forma en la atmósfera como consecuencia de diversos procesos, bajo la acción de la energía fotoeléctrica ((hv: energía fotoeléctrica):
NO2 + hv (menos de 310 nm) → O + NO
O + O2 → O3
En concreto, se requiere siempre radiación ultravioleta de longitud de onda menor de 242 nm, para descomponer la molécula diatómica de oxígeno, base de la formación de este gas. Se forma en grandes cantidades cuando tenemos una radiación ultravioleta considerable, y una alta concentración de nitrógeno, como la que puede llegar a darse sobre las ciudades como consecuencia de los fenómenos de contaminación urbana (no confundir con el ozono de las capas altas de la atmósfera). Se trata, por tanto de un contaminante de efectos oxidantes, y que por tanto es bastante agresivo con muchos seres vivos, en especial cuando se alcanzan determinadas concentraciones.
Los hidrocarburos: se forman como consecuencia de la combustión incompleta de gasolinas, y algunos tienen demostrados efectos cancerígenos: benceno, butadieno.
Los aldehídos: Son también contaminantes secundarios, originados a partir de los hidrocarburos formados en la combustión de gasolinas:
CH4 + 2 O2 + 2 NO → H2O + HCHO (formaldehído) + 2 NO2
Son moléculas irritantes, especialmente para los ojos.
La presencia de sales en la atmósfera ha de estar ligada, inevitablemente, a la presencia de abundante agua, en forma de aerosoles. En concreto, las más comunes son las sales de origen marino, en el aire cargado de gotitas de agua relacionado con la acción del oleaje. En el caso de la minería, es también relativamente frecuente la formación de aerosoles, ya sea como consecuencia de regado de pistas, o como consecuencia de voladuras en áreas no secas (muy poco común), o como consecuencia de riegos durante procesos de hidrometalurgia. Este último caso puede llegar a ser bastante problemático, pues durante estos procesos se emplean compuestos de alta toxicidad, como el cianuro de sodio (NaCN) en la pilas de lixiviación aurífera, o el ácido sulfúrico (H2SO4), para la lixiviación en pila de oxidados y sulfuros de cobre.
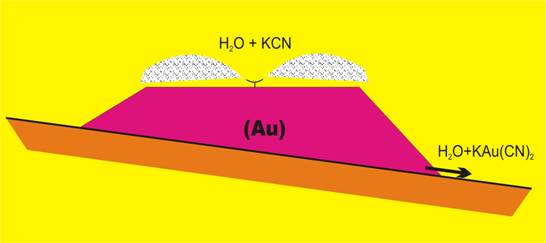
Esquema del proceso de lixiviación en pila.
8.6.- Bibliografía
Higueras, P., Oyarzun, R., Biester, H., Lillo, J. y Lorenzo, S. 2003. A first insight into mercury distribution and speciation in the Almadén mining district, Spain. Journal of Geochemical Exploration, 80: 95-104.
Krauskopf, K.B., y Bird, D.K. 1995. Introduction to Geochemistry. MacGraw-Hill, NY, 647 pp.
López García, J.A. 2004. Gossans. http://www.ucm.es/info/crismine/gossan/ gossanapuntes2.htm
Scheiner, B.J., Doyle, F.M., y Kawatra, S.K. (Eds.). 1989. Biotechnology in Minerals and metal processing. Society of Mining Engineers Inc., Littleton (CO), 209 pp.
Smith, J.V. (Ed.) 1999. Colloquium on geology, mineralogy, an human welfare. National Academy of Sciences, Washington DC, 96: 3348-3485.
Tarbuck, E.J. y Lutgens, F.K. Ciencias de la Tierra. 2000. Una introducción a la geología física. Prentice Hall Iberia, Madrid. 563 pp.
Documentos web:
http://h2osparc.wq.ncsu.edu/info/wetlanda/wetloss.html
http://www.who.int/pcs/ehc/summaries/ehc_224.html
http://www.usgs.gov./themes/factsheet/146-00/
http://www.epa.nsw.gov.au/leadsafe/leadinf8.htm
http://www.ucm.es/info/crismine/gossan/gossanapuntes2.htm
http://www.science.macmaster.ca/Biology/4S03/HM1.HTM
http://www.cochilco.cl/content/b-sustent/nacional/emisiones.html
http://www.portofentry.com/Environment/Technology/arsenicmng.html
http://minerals.usgs.gov/minerals/pubs/commodity/cadmium/
http://www.ambio.kva.se/2000/Nr3_00/May00_5.shtml
http://www.who.int/inf-fs/en/fact238.html
http://www.hc-sc.gc.ca/english/iyh/environment/asbestos.html
9.- Minerales de aplicación medioambiental
Estructura en túneles de una zeolita.
Algunos minerales tienen propiedades que permiten que sean utilizados en el control de procesos ambientales, para aplicaciones diversas, como pueden ser la depuración de aguas o la inmovilización de contaminantes presentes en el suelo. En el presente tema analizaremos algunas de estas aplicaciones más importantes. Los aspectos concretos que vamos a analizar en este ámbito serán los siguientes:
– Depuración de agua por intercambio iónico
– Depuración de aguas y suelos mediante sorción
– Depuración de olores
Organización del Capítulo:
9.1.- Grupo de las zeolitas
9.2.- Minerales de la arcilla
9.2.1.- Estructura y clasificación de los minerales de arcilla
9.2.2.- La captación iónica (intercambio y procesos de adsorción)
9.2.3.- Absorción y retención de líquidos
9.3.- Minerales del grupo de la crandallita
9.4.- Óxidos y oxidróxidos de hierro y manganeso
9.5.- Los carbonatos
9.5.1.- Canales abiertos de caliza (open limestone channels)
9.5.2.- Pozos bifurcados (diversion wells)
9.5.3.- Drenaje anóxico en calizas (anoxic limestone drains)
9.5.4.- Reactores de flujo vertical (vertical flow reactors)
9.6.- Bibliografía
9.1.- Grupo de las zeolitas
“Rarely in our technological society does the discovery of a new class of inorganic materials result in such a wide scientific interest and kaleidoscopic development of applications as has happened with the zeolite molecular sieves” (Donald W. Breck)
Esta frase lo resume todo o casi todo sobre estos maravillosos materiales englobados genéricamente bajo el nombre de “zeolitas”, cuyas aplicaciones industriales y ambientales aun están en fase desarrollo continuo. Las zeolitas constituyen un grupo de silicatos con una gran capacidad de intercambio iónico. Estas poseen una estructura definida por un armazón principal y “túneles”, que les confiere una propiedad que denominaremos “porosidad”, y que las convierte en microtamices muy efectivos. El armazón estructural está constituido por tetraedros de Si-O (SiO44-) y Al-O (AlO45-) que se unen por los vértices. Dado que el silicio presenta típicamente valencia 4 (Si4+) y el aluminio valencia 3 (Al3+), las zeolitas se encuentran “descompensadas eléctricamente” por lo que necesitan incorporar cationes para mantener la neutralidad (de ahí sus propiedades de intercambio). Dichos cationes, agua, u otras moléculas se acomodan en las estructuras tipo túnel.
Dos estructuras zeolíticas, note los tetraedros y las estructuras tipo túnel (poros)
donde se acomodan los cationes (que compensan eléctricamente la estructura),
el agua y otras moléculas.
La carga negativa en las zeolitas es compensada por cationes del tipo N+, K+ o NH4+, los se disponen en el interior de la estructura. Estos cationes son móviles y susceptibles de ser intercambiados por otros. Una reacción típica de intercambio iónico sería la siguiente:
Na-zeolita + Ca2+ = Ca-zeolita + 2 Na+
Las zeolitas ocurren en la naturaleza como minerales, y pertenecen al grupo de los tectosilicatos. Existen unas 40 zeolitas naturales y más de 150 sintéticas. Algunas zeolitas naturales incluyen:
| Mineral | Fórmula |
| Natrolita | Na2(Al2Si3O10)·2H2O |
| Chabazita | (Ca,Na)2 (Al2Si4O12)·6H2O |
| Analcima | Na(AlSi2O6)·H2O |
| Stilbita | Ca(Al2Si7O18)·7H2O |
| Heulandita | Ca(Al2Si7O18)·6H2O |
| Laumontita | Ca(Al2Si4O12)·4H2O |
| Mesolita | Na2Ca2(Al2Si3O10)·3H2O |
| Thompsonita | NaCa2(Al,Si)10O20·6H2O |
En la actualidad, la mayor parte de las zeolitas que se emplean para aplicaciones industriales son sintéticas: se diseñan “a medida” de un determinado proceso, es decir, para favorecer la captación de un determinado ión y su posterior elución en condiciones controladas, y se sintetiza el compuesto zeolítico en un proceso industrial de bajo coste económico. Debido a sus propiedades porosas las zeolitas se emplean en una gran variedad de usos industriales, y se comercializan en un mercado mundial de varios millones de toneladas por año.
El comportamiento de intercambio iónico en las zeolitas depende de varios factores, que determinan su selectividad a determinados cationes:
– Naturaleza de los cationes: tamaño, carga iónica, forma
– Temperatura
– Concentración de los cationes en solución
– Aniones asociados con los cationes en solución
– Solvente (agua, solventes orgánicos)
– Estructura de la zeolita (topología de la red, densidad de carga de la red)
La capacidad de intercambio iónico de una zeolita está directamente relacionada con la cantidad de aluminio presente en su red cristalina y depende directamente de su composición química: una alta capacidad de intercambio iónico corresponde a las zeolitas con baja relación SiO2/Al2O3. La capacidad de intercambio iónico teórica máxima, número de equivalentes intercambiables por masa de la celda unitaria, no siempre puede ser alcanzada debido a la existencia de sitios de intercambio inaccesibles.
Todo hasta aquí es atribuible tanto a zeolitas sintéticas como a las naturales. Desde el punto de vista del control ambiental mediante la eliminación de contaminantes, la gran mayoría de los autores coinciden en la superioridad de las zeolitas naturales atendiendo a:
– bajo costo de extracción y acondicionamiento para el intercambio,
– disponibilidad de grandes volúmenes,
– excelente estabilidad a los procesos químicos y térmicos que permite su reactivación y
– posibilidad de utilización en varios ciclos.
No obstante, las posibilidades de diseño de las zeolitas sintéticas y su carácter de compuestos químicamente puros, frente a las variaciones que pueden presentar las zeolitas naturales, hacen que las sintéticas se empleen cada vez más.
Algunas aplicaciones ambientales de las zeolitas incluyen la substitución de fosfatos como ablandadores del agua (captación de Ca2+), la remoción de óxidos de nitrógeno (NOx), y una contención en las emisiones de compuestos orgánicos volátiles (VOCs: volatile organic compounds). Los óxidos de nitrógeno son un blanco típico de cualquier política ambiental mínimamente bien concebida. Los compuestos tipo NOx fomentan la formación de oxidantes fotoquímicos contaminantes como el ozono (O3). Los compuestos de óxidos de nitrógeno se producen por la actividad eléctrica atmosférica (rayos), acción bacteriana, y por los volcanes. Sin embargo, estos óxidos también tienen un origen industrial, principalmente en la combustión de combustibles fósiles a temperaturas superiores a 1000ºC. En este sentido las zeolitas pueden jugar un papel muy importante, ya que mientras los procesos de eliminación clásicos de los NOx por Reducción Selectiva Catalítica solo son efectivos a temperaturas de no más de 450º (Pt, V), las zeolitas pueden llegar hasta los 600ºC, alcanzándose así una mayor efectividad en el proceso. En cuanto a los VOCs, estos se encuentran presenten en las gasolinas, solventes orgánicos, desgrasadores, lacas, etc., y al igual que los NOx son potenciadores de la formación de oxidantes fotoquímicos. Un tema aun no resuelto del todo en los coches es el arranque del motor en frío. Durante los dos primeros minutos el convertidor catalítico (obligatorio en muchos países del mundo) no es efectivo, y los VOCs se arrojados a la atmósfera. En este momento se están investigando zeolitas que puedan actuar durante ese lapso de tiempo en el convertidor catalítico del coche: una vez que se alcanzara la temperatura adecuada, la zeolita liberaría los VOCs, y el convertidor podría realizar su trabajo de manera adecuada.
9.2.- Minerales de la arcilla
Bajo el nombre de minerales de la arcilla se engloban distintas especies minerales según el uso que se haga del término arcilla. En unos casos se refiere exclusivamente a los filosilicatos de grano fino producto de la meteorización de otros minerales silicatados; en su acepción más amplia, se refiere a todos los minerales, silicatados o no, que se forman como consecuencia de estos procesos, y que simplemente tienen tamaño de grano inferior a 2 micras (aproximadamente, porque distintos autores fijan este límite en otros valores, por ejemplo, 4 micras). En cualquier caso, cuando hablamos de arcillas un componente fundamental van a ser siempre los filosilicatos que se forman en los procesos de alteración, cuya composición mineralógica puede ser muy variada en el detalle, y que van a ir siempre acompañados por proporciones variables de otros minerales.
Las aplicaciones de estos minerales o de estas mezclas mineralógicas en el campo ambiental resultan variadas: hemos visto su aplicabilidad en el campo del aislamiento de residuos industriales, y aquí estudiaremos otras aplicaciones como la depuración de líquidos y la absorción de olores.
9.2.1.- Estructura y clasificación de los minerales de arcilla
Las arcillas consisten en apilamientos poliméricos tipo sandwich de capas de tetraedros y octaedros. Las capas tetraédricas (T) están compuestas de Si-O, mientras que las octaédricas (O) de Al-O y Al-(OH). Como veremos más adelante, el silicio puede ser substituido por aluminio en las capas tetraédricas, y el aluminio por cationes divalentes (Mg, Fe2+) en las octaédricas. Dependiendo de la organización espacial de las capas (TO-TO- …, TOT-TOT- …) clasificaremos a las arcillas en dos tipos: 1:1 y 2:1. El tipo 1:1 consiste en una capa tetraédrica unida a una octaédrica (TO). Arcillas representativas de este tipo son las del grupo de la caolinita, cuya fórmula genérica es:
[Sin1Al4-n1]Aln1 + n2-4Fen33+O10(OH)8·nH2O
donde n = 1, 2, 3. La capa octaédrica tiene solo dos tercios de los huecos ocupados (cationes trivalentes), razón por la cual hablaremos de una arcilla “dioctaédrica” (2 de cada 3 huecos octaédricos ocupados), con un coeficiente de hidratación (moléculas de agua) = 0, excepto para la halloysita, con nH2O = 4.
Estructura 1:1 (TO-TO- …) de arcillas tipo caolinita.
La estructura tipo 2:1 consiste en una capa octaédrica cubierta a la manera de un sandwich por dos capas tetraédricas (TOT). Tres grupos de arcillas presentan esta estructuración: illita, vermiculita y esmectita. Su fórmula química genérica es:
Cx[Sin1Al8-n1] Aln1+n2-8 Fen33+Fen42+Mgn5Mn6O20(OH)4
Cx representa x moles de un catión univalente (1+; por ejemplo, K en illita) que permite balancear la carga negativa creada por: 1) el reemplazo isomorfo de Si (4+) por Al (3+) en la capas tetraédricas; 2) el reemplazo isomorfo de Al por Mg o Fe2+ en las capas octaédricas; o 3) el reemplazo de Mg en los octaedros por un catión dado, por ejemplo, Li en la hectorita.
Estructura 2:1 (TOT-TOT- …) de arcillas
tipo esmectita.
Los oxígenos de las capas tetraédricas que se disponen arriba y abajo de la octaédrica en las estructuras tipo 2:1, se distribuyen formando hexágonos con un hueco central. Si se produce una substitución en la capa octaédrica de Al por Mg o Fe2+, el exceso de cargas negativas hará que la superficie de oxígenos de los tetraedros adquiera una carga que permite ligar cationes (carga positiva) en los huecos anteriormente mencionados. Si además se producen substituciones de Si4+ por Al3+ en los tetraedros, la carga negativa, y por lo tanto capacidad de atracción de cationes se incrementará aún más.
También se desarrollan cargas negativas en los bordes del armazón de las arcillas, aunque su importancia disminuye del tipo 1:1 al 2:1.
9.2.2.- La captación iónica (intercambio y procesos de adsorción)
La capacidad de captación iónica varía de una arcilla a otra y se relaciona directamente con las características estructurales antes señaladas. En el tipo 1:1 el armazón TO-TO-…. se sustenta mediante enlaces tipo puente de hidrógeno, entre los grupos (OH) de las capas octaédricas y los oxígenos de las tetraédricas. Este enlace es lo suficientemente fuerte como para mantener la estructura global y al mismo tiempo impedir la entrada de cationes entre capas. Por el contrario, en las arcillas 2:1 este tipo de enlace entre capas no es posible (se enfrentan capas tetraédricas). En este caso el armazón estructural se compensa eléctricamente introduciendo cationes interlaminares, por ejemplo K en la illita. En las esmectitas se producen múltiples substituciones en las capas octaédricas y tetraédricas, que le confieren al igual que a la illita una carga global negativa. Sin embargo, esta carga es sólo un tercio de la de la illita, razón por la cual no se pueden ligar cationes de una manera fuerte en el espacio interlaminar. De esta manera, se permite la entrada de agua y otras moléculas que causan un aumento en el espacio interlaminar (“hinchamiento” de la arcilla). Con el agua vienen cationes disueltos, que su vez, pueden ser fijados en dicho espacio.
Así, las capacidades sorcitivas de las arcillas en general, y del grupo de la esmectita en particular, provienen de una paradoja estructural: las arcillas están “mal hechas”, presentando tal descompensación de cargas que justamente por eso, pueden captar cationes o moléculas. Dicho en otras palabras, si las arcillas no estuvieran descompensadas eléctricamente, su interés como intercambiadores iónicos sería nulo. De esta manera, y como corolario humorístico, podríamos añadir que una chapuza de la naturaleza (trabajo mal hecho) no tiene por qué tener, necesariamente, nefastas consecuencias.
Así, la capacidad de intercambio iónico es la propiedad fundamental de las arcillas del grupo de la esmectita, las que son capaces de adsorber con gran facilidad, los iones fijados en la superficie exterior de sus cristales, en los espacios interlaminares, o en otros espacios interiores de las estructuras.
Resumiendo, la capacidad de intercambio catiónico se puede definir como la suma de todos los cationes de cambio que un mineral puede adsorber a un determinado pH. Es equivalente a la medida del total de cargas negativas del mineral. Estas cargas negativas pueden ser generadas de tres formas diferentes:
* Sustituciones isomorfas dentro de la estructura.
* Enlaces insaturados en los bordes y superficies externas.
* Disociación de los grupos hidroxilos accesibles.
El primer tipo es conocido como carga permanente y supone un 80 % de la carga neta de la partícula; además es independiente de las condiciones de pH y actividad iónica del medio. Los dos últimos tipos de origen varían en función del pH y de la actividad iónica, corresponden a bordes cristalinos químicamente activos, y representan el 20 % de la carga total de la lámina.
A continuación se muestran algunos ejemplos de capacidad de intercambio catiónico (en meq/100 g):
| Caolinita | 3-5 |
| Halloisita | 10-40 |
| Illita | 10-50 |
| Clorita | 10-50 |
| Sepiolita-palygorskita | 20-35 |
| Montmorillonita | 80-200 |
| Vermiculita | 100-200 |
9.2.3.- Absorción y retención de líquidos
Otra aplicación común de algunos minerales arcillosos, en concreto de los de los grupos de sepiolita-palygorskita y esmectitas es como absorbentes de líquidos: son capaces de absorber agua u otras moléculas en el espacio interlaminar (esmectitas) o en los canales estructurales (sepiolita y paligorskita). Desde el punto de vista medioambiental, estos líquidos pueden ser contaminantes (por ejemplo, vertidos de hidrocarburos), o pueden contener contaminantes en disolución. Estas arcillas especiales se forman bajo condiciones climáticas muy específicas, o a partir de rocas de composición muy determinada, y que por sus características especiales presentan unas características que las hacen de gran utilidad en trabajos de descontaminación por su capacidad de adsorción e intercambio iónico.
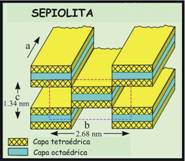
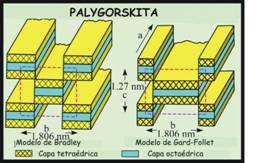
Estructura fundamental de la sepiolita y paligorskita. Note los canales tipo túnel.
La capacidad de intercambio iónico de las esmectitas ha promovido una serie de investigaciones conducentes a su utilización como agente catalítico, los que tienen un alto potencial como adsorbentes para tratar aguas o suelos contaminados. Los minerales del grupo de las esmectita incluyen arcillas dioctaédricas tales como la montmorillonita, beidellita, y natronita, y trioctaédricas como la hectorita (rica en Li), la saponita (rica en Mg), y la sauconita (rica en Zn).
El “problema” de las arcillas esmectíticas radica en que a temperaturas por encima de 200ºC la estructura colapsa, eliminando la zona de adsorción, de forma que si queremos depurar soluciones a temperaturas por encima de 200ºC no tendremos el espacio interlaminar donde acomodar los cationes o moléculas que nos interesa eliminar de la solución. La respuesta a este problema ha viene de un proceso denominado “pilarización”, que, como su nombre sugiere, consiste en intercalar “pilares” entre las capas tetraédricas para evitar el colapso estructural del mineral. Uno de los agentes de pilarización más comunes es el catión polinuclear de hidroxialuminio Al13O4(OH)283+, el cual es estable hasta temperaturas por encima de 500ºC.
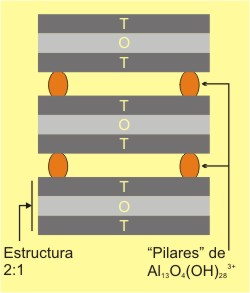
Pilarización de arcilla tipo 2:1 (TOT-TOT).
Este proceso tiene una gran ventaja: al introducir elementos pilarizantes de distintos tamaños podemos modificar el especiado entre las capas tetraédricas “a la medida” del tamaño de las partículas o iones que queremos adsorber.
Otro tratamiento posible para las arcillas esmectíticas consiste en la intercalación de cationes cuaternarios de amonio, los cuales interactúan con las arcillas reemplazando los cationes intercambiables desde su superficie. Como resultado del mayor tamaño de los cationes cuaternarios de amonio la distancia interlaminar aumenta, lo cual facilita la atracción de compuestos orgánicos (comportamiento organofílico) del tipo BTEX: benceno, tolueno, etilbenceno, y xileno, la fracción aromática peligrosa de las gasolinas.
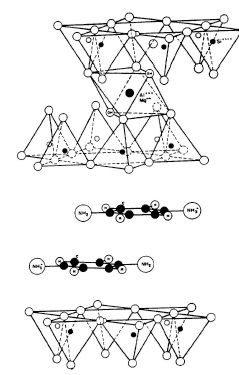
Intercalación cationes cuaternarios de amonia
para incrementar la distancia interlaminar
en una arcilla esmectítica y favorecer la incorporación de
compuestos orgánicos tipo BTEX.
Por su parte, la sepiolita y palygorskita (arcillas fibrosas) poseen estructuras cristalinas tipo túnel, que pueden albergar cationes y moléculas de agua por intercambio iónico con Ca y Mg, y agua zeolítica. Es en estas estructuras tipo túnel donde pueden acumular los contaminantes.
9.3.- Minerales del grupo de la crandallita
El grupo de la crandallita es un grupo mineralógico no muy conocido, caracterizado por el mineral que le da nombre: CaAl3(PO4)2(OH)5·(H2O).

Crandallita.
Los minerales del grupo de la crandallita son muy variados químicamente, aunque tienen en común el incluir grupos [PO4]. Algunos de estos minerales son:
| Mineral | Fórmula |
| Arsenocrandallita | (Ca,Sr)Al3[(As,P)O4]2(OH)5·(H2O) |
| Arsenogorceixita | BaAl3AsO3(OH)(AsO4,PO4)(OH,F)6 |
| Arsenogoyazita | (Sr,Ca,Ba)Al3(AsO4,PO4)2(OH,F)5·(H2O) |
| Eylettersita | (Th,Pb)1-xAl3(PO4,SiO4)2(OH)6 |
| Gorceixita | BaAl3(PO4)(PO3OH)(OH) |
| Goyazita | SrAl3(PO4)2(OH)5·(H2O) |
| Philipsbornita | PbAl3(AsO4)2(OH)5·(H2O) |
| Plumbogummita | PbAl3(PO4)2(OH)5·(H2O) |
| Waylandita | BiAl3(PO4)2(OH)6 |
| Zairita | Bi(Fe,Al)3(PO4)2(OH)6 |
| Minerales del Grupo de la Crandallita | |
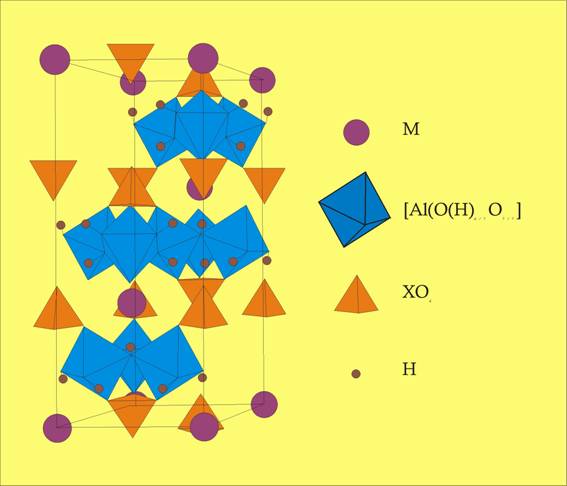
Estructura de los minerales del grupo de la crandallita
Esta amplia variabilidad composicional se traduce en una alta capacidad de intercambio iónico, en las tres posiciones estructurales que el grupo presenta (ver figura anterior): en los huecos tetraédricos de tipo XO4 (intercambiabilidad aniónica, de fósforo y/o azufre: por ejemplo el As puede entrar en estas posiciones, como arseniato), en los octaédricos (el aluminio puede ser sustituido por otros iones de carga y tamaño similares) y en las posiciones M, de coordinación 12 (el Ca y el Sr que normalmente ocupan estas posiciones pueden ser sustituidos por metales pesados como Hg, Pb, Cu, etc.). Esta propiedad hace que estos minerales puedan ser utilizados en la inmovilización de metales pesados de medios acuosos, aplicación que aún está ya desarrollada y patentada para el caso de elementos radioactivos del grupo de las Tierras Raras, y está en fase de estudio para otros metales pesados con mala consideración ambiental.
9.4.- Óxidos y oxidróxidos de hierro y manganeso
Otros minerales que debido a sus propiedades coloidales y estructurales poseen una gran importancia en los procesos naturales (y ahora industriales), en la “fijación” de metales de alta toxicidad, son los óxidos y oxidróxidos de hierro y manganeso.
Entre los óxidos y oxidróxidos de hierro destacaremos las siguientes fases minerales:
- Ferrihidrita, Fe5HO8•4H2O (también expresada como Fe(OH)3), la que se transforma a:
- Hematites, α-Fe2O3
- Goethita, α-FeO(OH)
Otros óxidos u oxidróxidos de hierro incluyen la lepidocrocita (γ-FeO(OH)) y la magnetita (Fe3O4), la cual a su vez puede dar lugar por oxidación (parte del hierro en la magnetita está como Fe2+) a maghemita (γ-Fe2O3). Los oxidróxidos de hiero se comportan como coloides positivos, y como tales tienen la capacidad de fijar por adsorción a aniones y aniones complejos. Entre estos debemos destacar por su alta peligrosidad dos especies:
- CrO42–, en otras palabras: Cr6+
- Aniones complejos de As (H2AsO4–, AsO43–, entre otros)
De esta manera los suelos o sedimentos fluviales con oxidróxidos de hierro poseen la capacidad natural de depurar soluciones (naturales o industriales) con productos tóxicos de este tipo. Aunque su presencia sea escasa, reconoceremos la presencia de fases oxidadas de hierro por el color rojo-anaranjado que le dan a los suelos o sedimentos.

Aspecto de sedimentos fluviales con oxidróxidos de hierro. Río Toro, Chile.

Captación de arsénico (sectores claros) en goethita. Río Toro, Chile. Imagen SEM-EDX.
En cuanto a los tratamientos industriales de aguas podemos destacar dos tecnologías:
- La oxidación de FeSO4 a fases del tipo FeO(OH) para la adsorción de especies acuosas de arsénico (aniones complejos de arsenito o arsenato).
- Procesos redox que implican la oxidación de Fe0 a Fe3+ y la reducción en paralelo de Cr6+ a Cr3+.
Las fases oxidadas de manganeso han sido menos tratadas en la literatura sobre temas ambientales, aunque también presentan un interesante potencial como depuradoras naturales o sintéticas. Dado que los óxidos de manganeso presentan dos estados de valencia (3+, 4+) la variedad de especies minerales oxidadas es mayor que la de los de hierro, que solo presentan el estado 3+. Otra diferencia con las especies oxidadas de hierro radica en el comportamiento de las partículas coloidales, las cuales presentan en este caso una carga neta negativa. Sin embargo, al igual que en el caso del hierro, también encontramos fases hidratadas (e.g. birnessita: (Ca,Na)(Mn2+,Mn4+)7 O14•3H2O) o de oxidróxidos (e.g. manganita: γ-MnO(OH)), así como de óxidos en el sentido estricto (e.g. pirolusita: β-MnO2).
A efectos de este capítulo destacaremos cuatro fases minerales:
| Mineral | Fórmula |
| Criptomelana | K1-2(Mn2+,Mn4+)8O16•xH2O |
| Todorokita | (Na,Ca,K)(Mn2+,Mg)Mn4+O12•xH2O |
| Litioforita | (Al,Li)(Mn2+,Mn4+)O2(OH)2 |
| Birnessita | (Ca,Na)(Mn2+,Mn4+)7O14•3H2O |
| Óxidos de manganeso | |
Estas fases oxidadas están basadas en un armazón estructural constituido por unidades de octaedros [MnO6], los que comparten vértices o aristas. El armazón tipo criptomelana y todorikita presenta túneles estructurales, mientras que el de la litioforita y birnessita se presenta en capas.

Estructura tipo túnel en un óxido sintético de manganeso (Na0.44MnO2)
mostrando octaedros de MnO6, y átomos de sodio en los túneles.
Una estructura como la de la todorokita puede admitir cationes tales como Co2+, Ni2+ y Cu2+. Este mineral posee obviamente capacidades de intercambio iónico, primero adsorbiendo, y luego estabilizando los cationes. En las estructuras en capas (estratificadas) la admisión de cationes (e.g. Co2+) seguiría los siguientes pasos: 1) adsorción inicial de Co2+ cerca de las vacancias en la capa de unidades [MnO6]; oxidación de Co2+ a Co3+; y 3) ocupación final de los sitios octaédricos por Co3+.
A escala de laboratorio se ha experimentado con mezclas de pirolusita-goethita (Mn:Fe ~ 10:1) para la descontaminación de aguas arseniacales. Aunque los resultados han sido positivos, no queda claro el como y el porqué de este fenómeno. Recordemos que las especies acuosas de As son aniones complejos (carga negativa), y que también la carga de las partículas coloidales de las fases oxidadas de manganeso es negativa!!! Resulta difícil así visualizar como el arsénico podría ser adsorbido, salvo que no sea la pirolusita sino la goethita la fase mineral que efectivamente fija el arsénico.
9.5.- Los carbonatos
Los carbonatos constituyen un grupo mineralógico importante por su variedad, y por la abundancia de determinados minerales del grupo, como la calcita o la dolomita, que constituyen las calizas y dolomías, respectivamente. Presentan además aplicaciones variadas, sobre la base de: 1) su abundancia y el bajo coste de su extracción minera; 2) el hecho de reaccionar con los ácidos y otros compuestos químicos, como la sílice (dando origen al cemento) y el dióxido de azufre (dado origen a yeso/anhidrita); y 3) porque su descomposición térmica se produce a temperatura relativamente baja (del orden de los 600ºC) dando origen a la denominada cal viva (CaO) o a la cal dolomítica (CaO+MgO), que a su vez tienen aplicaciones importantes. Dentro del campo medioambiental, las calizas tienen aplicación fundamentalmente en procesos de desulfuración durante procesos de combustión o metalurgia, procesos ambos en los que a menudo se producen óxidos de azufre gaseosos que escapan al medio ambiente, dando origen al fenómeno de lluvia ácida. La presencia de caliza en el medio de combustión, ya sea como lecho fluido, o como aditivo, impide la formación de estos compuestos:
CaCO3 + SO2 + 2 H2O + 0.5 O2 → CaSO4 · 2 H2O + CO2
La adición de caliza a los suelos origina los siguientes procesos: 1) neutralización de la acidez; 2) incremento de la saturación en bases (Ca) del suelo; 3) cambios en los ratios de cationes básicos absorbidos y disueltos; 4) incremento del pH del suelo, lo cual afecta, además, a la solubilidad de varios compuestos; 5) se produce la inactivación de concentraciones tóxicas de Al, Mn y otras sustancias; 6) la fijación de nitrógeno y la mineralización aumenta, al incrementarse el pH y la saturación en bases; 7) la concentración de electrolitos aumenta con la disolución de la caliza.
Uno de los mayores problemas que plantea la minería es el drenaje ácido (DAM). Para su tratamiento se pueden emplear diversas técnicas, aunque aquí nos centraremos en aquellas en se emplean rocas calcáreas, como las calizas (constituidas principalmente por calcita: CaCO3). En este caso el carbonato de calcio actúa como un reactivo neutralizante que lleva al pH a valores aceptables, y favorecen la precipitación de la mayor parte de los metales pesados que pueda contener el agua. El proceso que consume los hidrogeniones de la solución ácida, y por lo tanto la neutraliza, lo podemos resumir de la siguiente manera:
CaCO3 + H+ → Ca2+ + HCO3–
Existen una serie de procedimientos industriales para conseguir dicha neutralización entre los que destacaremos los siguientes:
9.5.1.- Canales abiertos de caliza (open limestone channels)
Constituyen la forma más simple de tratar el DAM, y pueden ser de dos tipos: canales recubiertos de caliza a través de los cuales se hace pasar el agua a tratar, o simplemente, añadir trozos de caliza a los canales de desagüe ya existentes. El principal problema que pueden presentar es el de que los cantos de caliza se recubren de una lámina de óxidos e hidróxidos de hierro que los aíslan, reduciendo la efectividad del proceso a medio-largo plazo. Eso hace necesario utilizar grandes cantidades de caliza. Es también importante la impermeabilización del fondo del canal, para evitar la infiltración del DAM.
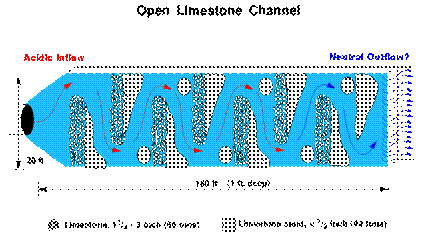
Canal abierto de caliza. Note a la izquierda como ingresa
la solución ácida (acidic inflow), y por la izquierda sale una solución
neutralizada (neutral outflow).
9.5.2.- Pozos bifurcados (diversion wells)
Es otra forma de tratar el DAM con caliza, la que se realiza en un “pozo” con circulación forzada de agua donde se acumula la caliza. La turbulencia del régimen y la presencia de partículas finas y abrasivas dificultan la formación de revestimientos aislantes en la caliza.
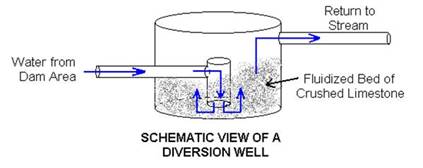
Esquema del proceso de pozo bifurcado. Por la izquierda ingresa la solución ácida,
que reacciona con el lecho de caliza triturada, escapando por la derecha como solución neutralizada.
9.5.3.- Drenaje anóxico en calizas (anoxic limestone drains)
Se trata de un sistema para interceptar y neutralizar flujos subterráneos de DAM, evitando además su contacto con el oxígeno atmosférico, lo que evita la oxidación de los metales, y por tanto, la formación de revestimientos de óxidos de Fe en la caliza.
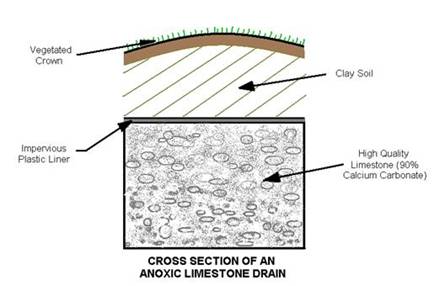
El sistema de drenaje anóxico.
9.5.4.- Reactores de flujo vertical (vertical flow reactors)
Consisten en celdas de tratamiento con una base de caliza y drenaje basal sobre la que se sitúa una capa de sustrato orgánico y una lámina de agua estática. El agua fluye verticalmente a través del compost y de la caliza, y se recoge y descarga a través de un sistema de tuberías. Este sistema incrementa la alcalinidad mediante la disolución de caliza y la reducción bacteriana de sulfatos. A continuación se requiere un tratamiento adicional, como puede ser en una laguna aeróbica, para la oxidación y precipitación de los metales pesados.
Esquema del sistema de flujo vertical.
9.6.- Bibliografía
Andrews, J.E., Brimblecombe, P., Jickells, T.D., y Liss, P.S. 1996. An introduction to environmental chemistry. Blackwell Science, Oxford, 209 pp.
Arnold, D.A. 1993. Porous manganese oxides. http://www.ri.ac.uk/DFRL/D.Arnold/
Chakravarty, S., Dureja, V., Bhattacharyya, G., Maity, S., y Bhattaacharjee, S. 2002. Removal of arsenic from groundwater using a low cost ferruginous manganese ore. Water Research, 36: 625-632.
Crespo, A., Lunar, R., Oyarzun, R., Doblas, M. 1995. Unusual case for hot-springs related Co-rich Mn mineralization in central Spain: the Pliocene Calatrava deposits. Economic Geology, 90: 433-437.
Gitipour, S., Bowers, MHuff, W., y Bodocsi, A. 1997. The efficiency of modified bentonite clays for removal of aromatic organics from oily liquid wastes. Spill Science and Technology Bulletin, 4: 155-164.
Marcus, B.K., y Cormier, W.E. 2003. Going green with zeolites. http://www.zeolyst. com./html/cep.html.
Smedley, P.L., y Kinniburgh, D.G. 2002. A review of the source, behavior and distribution of arsenic in natural waters. Applied Geochemistry, 17: 517-568.
Smith, J.V. (Ed.) 1999. Colloquium on geology, mineralogy, an human welfare. National Academy of Sciences, Washington DC, 96: 3348-3485.
Stip, S.L.S.; Hansen, M., Kristensen., Hochella, M.F., Bennedsen, L., Dideriksen, K., Balic-Zunic, T., Léonard, D., y Mathieu, H.J. 2002. Behavior of Fe-oxides relevant to contaminant uptake in the environment. Chemical Geology, 190: 321-337.
USGS 2003. A laboratory manual for X-Ray powder diffraction. http://pubs.usgs.gov/of/of01-041/htmldocs/clays/.
[/vc_column_text][/vc_tta_section][/vc_tta_tour][/vc_column][/vc_row]