Guanidinium iodide salts as single component catalysts for CO2 to epoxide fixation.
In this study, we present the synthesis, characterization and catalytic reactions of a new family of one-component catalysts based on guanidinium salts. The synthesis of cyclic carbonates from epoxides and CO2 at 70-80 degrees C under 1-5 bar of CO2 pressure is demonstrated using the new one-component guanidinium salts with various substituents. The 1e catalytic system, which has isopropyl-type substituents in its structure, has the best conversion rates; the TON for the synthesis of styrene carbonate was 92. The production of cyclic carbonates from the internal epoxides is another application for these catalytic systems with the conversion of cyclohexane oxide to the respective cyclic carbonate at 86% with 1e catalyst. Finally, ESI-MS was used to examine the interaction of the new organocatalysts with CO2 and epichlorohydrin. These findings led to catalysts made of organic molecules that are suitable for organocatalysis since they do not require a co-catalyst and can operate effectively at moderate pressures and temperatures.
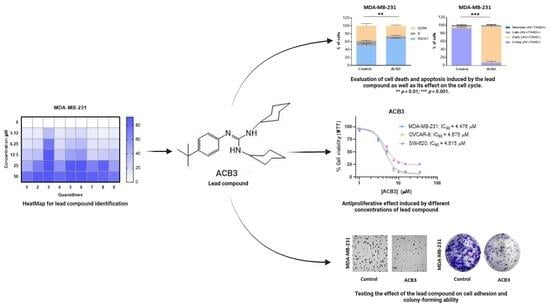
Guanylation Reactions for the Rational Design of Cancer Therapeutic Agents
Int. J. Mol. Sci. 2023, 24(18), 13820; https://doi.org/10.3390/ijms241813820
The modular synthesis of the guanidine core by guanylation reactions using commercially available ZnEt2 as a catalyst has been exploited as a tool for the rapid development of antitumoral guanidine candidates. Therefore, a series of phenyl-guanidines were straightforwardly obtained in very high yields. From the in vitro assessment of the antitumoral activity of such structurally diverse guanidines, the guanidine termed ACB3 has been identified as the lead compound of the series. Several biological assays, an estimation of AMDE values, and an uptake study using Fluorescence Lifetime Imaging Microscopy were conducted to gain insight into the mechanism of action. Cell death apoptosis, induction of cell cycle arrest, and reduction in cell adhesion and colony formation have been demonstrated for the lead compound in the series. In this work, and as a proof of concept, we discuss the potential of the catalytic guanylation reactions for high-throughput testing and the rational design of guanidine-based cancer therapeutic agents.
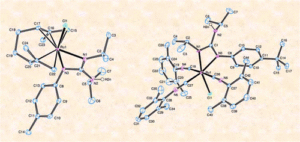
Imidazol(in)ium-2-Thiocarboxylate Zwitterion Ligands: Structural Aspects in Coordination Complexes
Crystals 2023, 13(9), 1304; https://doi.org/10.3390/cryst13091304
Azolium-2-thiocarboxylate zwitterion ligands have emerged as a promising class of compounds in the field of coordination chemistry due to their unique structural features and versatile applications. These ligands are characterized by a positively charged azolium ring and a negatively charged thiocarboxylate moiety, making them capable of forming stable coordination complexes with various metal ions. One of the key structural aspects that make these ligands attractive for coordination chemistry is their ability to adopt diverse coordination modes with metal centers. The nature of these ligands enables them to engage in both monodentate and bidentate coordination, resulting in the formation of chelated complexes with enhanced stability and controlled geometry or the formation of polynuclear structures. This versatility in coordination behavior allows for the design of tailored ligands with specific metal-binding preferences, enabling the creation of unique and finely tuned coordination architectures. The azolium-2-thiocarboxylate zwitterionic ligands offer a promising platform for the design of coordination complexes with diverse structural architectures.
ZnEt2 as a Precatalyst for the Addition of Alcohols to Carbodiimides
We report here the use of commercially available ZnEt2 as an efficient precatalyst for the addition of alcohols to carbodiimides to obtain a wide range of isoureas under mild conditions. In an initial screening using methanol and commercial carbodiimides as substrates, the bulky isourea (OMe)(NHDipp)C(NDipp) (Dipp = 2,6-iPr2C6H3) was prepared for the first time using a catalytic method, and its structure confirmed by an X-ray diffraction analysis. Then, the efficiency of the precatalyst was tested with two carbodiimides, C(NiPr)2 and C(Np-tol)2, toward a series of alkylic and arylic alcohols and diols, with different steric and electronic properties, including the presence of other functional groups, usually with excellent conversions, especially for the more reactive aromatic carbodiimide. Some of the new isoureas thus prepared have also been isolated and characterized. Kinetic and stoichiometric experiments allowed us to propose a plausible mechanism for these transformations.

Guanidinates as Alternative Ligands for Organometallic Complexes.
Molecules 2022, 27, 5962-5993.
https://doi.org/10.3390/molecules27185962.
For decades, ligands such as phosphanes or cyclopentadienyl ring derivatives have dominated Coordination and Organometallic Chemistry. At the same time, alternative compounds have emerged that could compete either for a more practical and accessible synthesis or for greater control of steric and electronic properties. Guanidines, nitrogen-rich compounds, appear as one such potential alternatives as ligands or proligands. In addition to occurring in a plethora of natural compounds, and thus in compounds of pharmacological use, guanidines allow a wide variety of coordination modes to different metal centers along the periodic table, with their monoanionic chelate derivatives being the most common. In this review, we focused on the organometallic chemistry of guanidinato compounds, discussing selected examples of coordination modes, reactivity and uses in catalysis or materials science. We believe that these amazing ligands offer a new promise in Organometallic Chemistry.
Aerobic/Room-Temperature-Compatible s-Block Organometallic Chemistry in Neat Conditions: A Missing Synthetic Tool for the Selective Conversion of Nitriles into Asymmetric Alcohols
ChemSusChem, 2022,15, e202201348(1of7
https://doi.org/10.1002/cssc.202201348
Highly-efficient and selective one-pot/two-step modular double addition of different highly polar organometallic reagents (RLi/RMgX) to nitriles en-route to asymmetric tertiary alcohols (without the need for isolation/purification of any halfway reaction intermediate) has been studied, for the first time, in the absence of external/additional organic solvents (neat conditions), at room temperature and under air/moisture (no protecting atmosphere is required); reaction conditions that are generally forbidden in the field of highly-reactive organolithium/organomagnesium reagents. Our one-pot modular tandem protocol demonstrated high chemoselectivity with a broad range of nitriles, as no side reactions (Li/Halogen exchange, ortho-lithiations or benzylic metalations) were detected. Finally, we were able to scale up this protocol, thus proving that our environmentally-friendly methodology is amenable for a possible applied synthesis of asymmetric tertiary alcohols under bench type reaction conditions and in the absence of external organic solvents.

Combination of air/moisture/ambient temperature compatible organolithium chemistry with sustainable solvents: selective and efficient synthesis of guanidines and amidines.
Green Chem., 2022, 24, 800-812. https://pubs.rsc.org/en/content/articlelanding/2022/gc/d1gc03393j
Highly-efficient and selective fast addition of in situ generated lithium amides [LiN(H)R] (obtained via an acid–base reaction between n-BuLi and the desired primary amine) into carbodiimides (R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN–R) or nitriles (R–C
CN–R) or nitriles (R–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) has been studied, for the first time, in 2-MeTHF or CPME as ethereal green solvents, at room temperature and in the absence of a protecting atmosphere (i.e., under air/moisture), reaction conditions that are generally forbidden in the field of highly-reactive main group polar compounds, such as lithium amides. The correct selection of: (i) the employed synthetic methodology (one-pot/two-step protocols are preferred); (ii) the unsaturated organic electrophile (carbodiimides or nitriles were assayed); and (iii) the sustainable ethereal solvent (2-MeTHF or CPME) allows the straightforward and successful synthesis of either guanidines or amidines under air/moisture at room temperature without the need for isolation or purification of any halfway reaction intermediate. Finally, we were able to scale up the reaction, thus proving that our new and environmentally-friendly protocol is amenable for a possible applied synthesis of highly-substituted iminic-type fine chemical products (guanidines/amidines) under bench-type reaction conditions.
N) has been studied, for the first time, in 2-MeTHF or CPME as ethereal green solvents, at room temperature and in the absence of a protecting atmosphere (i.e., under air/moisture), reaction conditions that are generally forbidden in the field of highly-reactive main group polar compounds, such as lithium amides. The correct selection of: (i) the employed synthetic methodology (one-pot/two-step protocols are preferred); (ii) the unsaturated organic electrophile (carbodiimides or nitriles were assayed); and (iii) the sustainable ethereal solvent (2-MeTHF or CPME) allows the straightforward and successful synthesis of either guanidines or amidines under air/moisture at room temperature without the need for isolation or purification of any halfway reaction intermediate. Finally, we were able to scale up the reaction, thus proving that our new and environmentally-friendly protocol is amenable for a possible applied synthesis of highly-substituted iminic-type fine chemical products (guanidines/amidines) under bench-type reaction conditions.
Novel Fluorescence Guanidine Molecules for Selective Sulfate Anion Detection in Water Complex Samples over a Wide pH Range.
ACS Sens. 2021, 6, 9, 3224-3233. https://pubs.acs.org/doi/abs/10.1021/acssensors.1c00876
Quantitative analysis of sulfate anions in water still remains an important challenge for the society. Among all the methodologies, the most successful one is based on optical supramolecular receptors because the presence of small concentrations of sulfate anion modifies the photophysical properties of the receptor. In this case, fluorescence anion sensors have been designed by the incorporation of guanidine motifs into fluorenyl cores. The photophysical behaviors of the new mono- (M) and bis-guanidine (B) derivatives were studied through pH dependence, solvent effects, and ion sensing on steady-state spectra and time-resolved fluorescence spectroscopy. In more detail, the results demonstrate that M is a highly selective and sensitive sulfate ion receptor in real water samples and, even more importantly, its function remains unchanged at different ranges of pH. The reason behind this resides on the fluorescence quenching produced by an internal charge-transfer process when the sulfate anion is complexed with M. It is worth noting that the global and partial affinity constants (1010 M–2 and 105 M–1, respectively) of complex formation are far above from the current sulfate sensors in water (104 M–1) which give an LOD of 0.10 μM in water with an analytical range of 2.5–10 μM. On the other hand, although it would seem, at first sight, that the B derivate will be the most promising one, the possibility of having two simultaneous protonation states reduces the complex formation and, therefore, its sensitivity to sulfate anions. The results presented here offer the possibility of using a new molecule in water environments, which opens the door to infinite applications such as the detection of trace amounts of sulfate ions in food or water.

Mono- and dinuclear asymmetric aluminum guanidinates for the catalytic CO2 fixation into cyclic carbonates
Organometallics 2021, 40, 2859-2969. https://doi.org/10.1021/acs.organomet.1c00319
A set of trisubstituted guanidine ligands L1H2–L4H2 with general formula (iPrHN)2CNR (R = Ph (L1H2), R = 2,4,6-Me3C6H2(L2H2), R = p-BrC6H4(L3H2), R = (C5H4)Fe(C5H5), Fc (L4H2)) was employed to synthesize a family of mono- and dinuclear asymmetric methyl aluminum guanidinato compounds ((L2H)AlMe2 (1), (L4H)AlMe2 (2), (L1)Al2Me4 (3), (L2)Al2Me4 (4), (L3)Al2Me4 (5), (L4)Al2Me4 (6), (L1H)2AlMe (7), (L2H)2AlMe (8), and (L4H)2AlMe (9)) that were characterized by NMR spectroscopy (1–9) and single-crystal X-ray diffraction (4 and 8). These compounds were tested as catalysts for the fixation of carbon dioxide with epoxides to give cyclic carbonates, using tetrabutylammonium iodide (TBAI) as cocatalyst. The reactions were performed under solvent-free conditions at 70 °C and 1 bar CO2 pressure. Complexes 1–9 were more active than their respective free guanidines under the same experimental conditions for the synthesis of styrene carbonate (11a). The dinuclear complex 6 was the most efficient and active catalyst for the synthesis of several monosubstituted carbonates (11a–l) with excellent conversions and selectivities. Furthermore, the formation of some disubstituted cyclic carbonates (13a–c) using this dinuclear aluminum catalyst was also studied.

New guanidine-borane adducts: An experimental and theoretical approach.
Inorg. Chim. Acta 2021, 518, 120217-120224. https://doi.org/10.1016/j.ica.2020.120217
We report here a series of new adducts incorporating arylguanidines Ar*NC(NiPrH)2 (Ar* = C6H5, (p-CN-C6H4), (p-CF3-C6H4) and (2,4,6-(CH3)3-C6H2)) bearing donor or acceptor substituents in the phenyl rings, and bispentafluorophenylborane [HB(C6F5)2]. The spectroscopic and structural features of the new compounds [(C6F5)2HBNAr*C(NiPrH)2] (Ar* = C6H5 1, (2,4,6-(CH3)3-C6H2) 2, (p-CN-C6H4) 3 and (p-CF3-C6H4) 4) have been studied by multinuclear NMR and X-ray diffraction techniques. The structural differences between 1 and 2–4 compounds have been explained using electronic structure descriptors based on DFT calculations at the B3LYP/6–31 + G(d,p) level in the gas phase.

Reactivity of N-Phosphinoguanidines of the Formula (HNR)(Ph2PNR)C(NAr) toward Main Group Metal Alkyls: Facile Ligand Rearrangement from N-Phosphinoguanidinates to Phosphinimine-Amidinates.
Inorganic Chemistry 2020, 59, 15262-15275.DOI: 10.1021/acs.inorgchem.0c02224
We report the reactivity of N-phosphinoguanidines of the formula (HNR)(Ph2PNR)C(NAr) (R = iPr and Ar = 2,6-iPr2C6H3 [Dipp] for 1a, R = iPr and Ar = 2,4,6-Me3C6H2 [Mes] for 1b, and R = Cy and Ar = Dipp for 1c), prepared in high yields from the corresponding trisubstituted guanidines, toward main group metal alkyls AlMe3, ZnEt2, MgnBu2, and nBuLi to obtain novel phosphinoguanidinato and phosphinimine-amidinato compounds. Reactions of 1a–c with AlMe3 at room temperature led to the kinetic phosphinoguanidinato products [Al{κ2–N,N′-(NR)C(NAr)(NRPPh2)}Me2] (2a–c), whereas the mild heating (60–80 °C) of solutions of 2a–c give the thermodynamic phosphinimine-amidinato products [Al{κ2–N,N′-(NR)C(NAr)(PPh2NR)}Me2] (3a–c) after ligand rearrangement. The reactions of equimolar amounts of 1a–c and ZnEt2 initially give solutions containing unstable phosphinoguanidinato compounds [Zn{κ2–N,P-(NR)C(NAr)(NRPPh2)}Et] (4a–c), which rearrange upon mild heating to the phosphinimine-amidinato derivatives [Zn{κ2–N,N′-(NR)C(NAr)(PPh2NR)}Et] (6a–c). Bis(phosphinoguanidinato) compounds [Zn{κ2–N,P-(NR)C(NAr)(NRPPh2)}2] (5a–c) can be obtained under mild conditions (<45 °C) in THF, whereas bis(phosphinimine-amidinato) compounds [Zn{κ2–N,N′-(NR)C(NAr)(PPh2NR)}2] (7a–c) are also accessible under more forcing conditions (55–100 °C) from (i) ZnEt2 and 1b,c (2 equiv), (ii) 6a and 1a, or (iii) 5b,c. Equimolar mixtures of MgnBu2 and 1a–c in THF at room temperature give unstable phosphinimine-amidinato monoalkyl products [Mg{κ2–N,N′-(NR)C(NAr)(PPh2NR)}nBu(THF)2] (8a–c), whereas 2 equiv of 1a,b are required to reach the bischelate compounds [Mg{κ2–N,N′-(NiPr)C(NAr)(PPh2NiPr)}2] (9a,b). Finally, phosphinoguanidinato compounds [Li{κ2–N,P-(NR)C(NDipp)(NRPPh2)}(THF)2] (10a,c) were obtained in the reactions of 1a,c with nBuLi in THF under ambient conditions. The removal of the solvent from solutions of 10a,c under partial vacuum leads to the dinuclear compounds [Li2{μ–κ2–N,N′:κ1–N–(NR)C(NDipp)(NRPPh2)}2(THF)2] (11a,c) after the decoordination of one of the THF molecules in 10a,c and dimerization. Heating solutions of 10a,c at 60 °C triggers ligand rearrangement to give phosphinimine-amidinato compounds [Li{κ2–N,N′-(NR)C(NDipp)(PPh2NR)}(THF)2] (12a,c). We also propose a mechanism for the ligand rearrangement reaction from 10a to give 12a, supported by DFT calculations, which fits nicely with our experimental results. It essentially involves a carbodiimide deinsertion reaction followed by a [3 + 2] cycloaddition between the resulting lithium phosphino-amide and the carbodiimide.

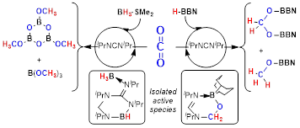
Ph2PCH2CH2B(C8H14) and Its Formaldehyde Adduct as Catalysts for the Reduction of CO2 with Hydroboranes
Inorganic Chemistry 2020, 59, 14, 9998-10012 DOI: 10.1021/acs.inorgchem.0c01152
We study two metal-free catalysts for the reduction of CO2 with four different hydroboranes and try to identify mechanistically relevant intermediate species. The catalysts are the phosphinoborane Ph2P(CH2)2BBN (1), easily accessible in a one-step synthesis from diphenyl(vinyl)phosphine and 9-borabicyclo[3.3.1]nonane (H-BBN), and its formaldehyde adduct Ph2P(CH2)2BBN(CH2O) (2), detected in the catalytic reduction of CO2 with 1 as the catalyst but properly prepared from compound 1 and p-formaldehyde. Reduction of CO2 with H-BBN gave mixtures of CH2(OBBN)2 (A) and CH3OBBN (B) using both catalysts. Stoichiometric and kinetic studies allowed us to unveil the key role played in this reaction by the formaldehyde adduct 2 and other formaldehyde–formate species, such as the polymeric BBN(CH2)2(Ph2P)(CH2O)BBN(HCO2) (3) and the bisformate macrocycle BBN(CH2)2(Ph2P)(CH2O)BBN(HCO2)BBN(HCO2) (4), whose structures were confirmed by diffractometric analysis. Reduction of CO2 with catecholborane (HBcat) led to MeOBcat (C) exclusively. Another key intermediate was identified in the reaction of 2 with the borane and CO2, this being the bisformaldehyde–formate macrocycle (HCO2){BBN(CH2)2(Ph2P)(CH2O)}2Bcat (5), which was also structurally characterized by X-ray analysis. In contrast, using pinacolborane (HBpin) as the reductant with catalysts 1 and 2 usually led to mixtures of mono-, di-, and trihydroboration products HCO2Bpin (D), CH2(OBpin)2 (E), and CH3OBpin (F). Stoichiometric studies allowed us to detect another formaldehyde–formate species, (HCO2)BBN(CH2)2(Ph2P)(CH2O)Bpin (6), which may play an important role in the catalytic reaction. Finally, only the formaldehyde adduct 2 turned out to be active in the catalytic hydroboration of CO2 using BH3·SMe2 as the reductant, yielding a mixture of two methanol-level products, [(OMe)BO]3 (G, major product) and B(OMe)3 (H, minor product). In this transformation, the Lewis adduct (BH3)Ph2P(CH2)2BBN was identified as the resting state of the catalyst, whereas an intermediate tentatively formulated as the Lewis adduct of compound 2 and BH3 was detected in solution in a stoichiometric experiment and is likely to be mechanistically relevant for the catalytic reaction.

Copper (II) as catalyst for intramolecular cyclization and oxidation of (1,4-phenylene)bisguanidines to benzodiimidazole-diylidenes
Journal of Catalysis, 2020, 382, 150-154. DOI: 10.1016/j.jcat.2019.12.002
A synthetically useful approach of catalytic intramolecular cyclization and oxidation of 2′,2′-(1,4-phenylene)bis(1,3-dialkyl)guanidines (Alkyl = isopropyl 1 or cyclohexyl 2) catalyzed by copper acetate in acetonitrile under air was studied by on line monitoring of the reaction by ESI-MS. All-important intermediates organic species were intercepted during the experiment confirming for the first time the stepwise (1,4-phenylene)bisguanidines cyclization and oxidation mechanism. Moreover, performed collision-induced dissociation (CID) experiments were also applied as a structure elucidation tool. Bimetallic copper intermediates Cu1 ([C28H48Cu2N6O10 + H]+) of m/z 755 and Cu2 [C22H36Cu2N6O4 + H]+ of m/z 575 were documented. The plausible key mechanistic steps involving the formation of organic and inorganic intermediates detected by in situ monitoring of the reaction are presented.

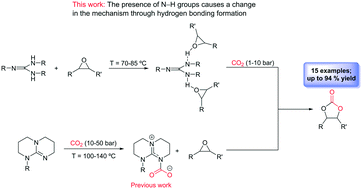
Aromatic guanidines as highly active binary catalytic systems for the fixation of CO2 into cyclic carbonates under mild conditions
Catalysis Science and Technology, 2019, 9, 3879-3886. DOI: 10.1039/C9CY00667B
We have synthesised a set of aromatic mono- and bis(guanidines) which are highly effective binary catalytic systems (guanidine/cocatalyst) for the formation of cyclic carbonates. The presence of multiple N–H bonds causes a modification in the traditional mechanism proposed for the synthesis of cyclic carbonates catalysed by guanidines through the formation of hydrogen bonds between the oxygen atom of the epoxide and the N–H groups of the guanidines. This change allows a considerable reduction of the reaction temperature and CO2 pressure employed in this process
9-Borabicyclo[3.3.1]nonane: a metal-free catalyst for the hydroboration of carbodiimides
Chem. Commun., 2019, 55, 3073. DOI: 10.1039/c9cc00593e
The commercial 9-borabicyclo[3.3.1]nonane dimer is used as the first example of a metal-free catalyst for the monohydroboration of carbodiimides with pinacol borane. Stoichiometric reactions, kinetic studies, and DFT calculations have allowed us to propose a plausible mechanism involving a heterocyclic amidinate intermediate with a three center-two electron B–H–B bond.
![Graphical abstract: 9-Borabicyclo[3.3.1]nonane: a metal-free catalyst for the hydroboration of carbodiimides](https://pubs.rsc.org/en/Image/Get?imageInfo.ImageType=GA&imageInfo.ImageIdentifier.ManuscriptID=C9CC00593E)
Unusual ligand rearrangement: from N-phosphinoguanidinato to phosphinimine-amidinato compounds
Chem. Commun., 2019, 55, 2809. DOI: 10.1039/c9cc00432g
Novel N-phosphinoguanidines (HNiPr)(Ph2PNiPr)C(NAr) (Ar = 2,6-iPr2C6H3, 2,4,6-Me3C6H2) react with AlMe3 to afford phosphinimine-amidinato derivatives, via an unprecedented rearrangement of an initial N-phosphinoguanidinato intermediate. A reasonable mechanism has been proposed for this transformation, supported by DFT calculations, involving carbodiimide de-insertion followed by a [3+2] cycloaddition.

Selective Three-Component Coupling for CO2 Chemical Fixation to Boron Guanidinato Compounds
Inorg. Chem., 2018, 57, 8404−8413 DOI: 10.1021/acs.inorgchem.8b01068
A selective three-component coupling was employed to fix carbon dioxide to boron guanidinato compounds. The one-pot reaction of carbon dioxide, carbodiimides, and borylamines (ArNH)BC8H14 afforded the corresponding 1,2-adducts {R(H)N}C{N(Ar)}(NR)-(CO2)BC8H14. Alternatively, the reaction with p-MeOC6H4NC or 2,6-Me2C6H3NC gave the corresponding isocyanide 1,1-adducts {i-PrHN}C{N(p-Me-C6H4)}(Ni-Pr)-{CNAr}BC8H14. The molecular structures of products (2,6-i-Pr2C6H3NH)BC8H14 7, {i-Pr(H)N}C{N(p-MeC6H4)}(Ni-Pr)(CO2)BC8H14 9, {Cy(H)N}C{N(p-MeC6H4)}(Cy)-(CO2)BC8H14 13, and {i-PrHN}C{N(p-MeC6H4)}(Ni-Pr)-{CNR″}BC8H14 (R″ = p-MeOC6H4, 2,6-Me2C6H3) 14 and 15 were established by X-ray diffraction. Density functional theory calculations at the M05-2X level of theory revealed that CO2 fixation and formation of the corresponding adduct is exothermic and proceeds via a nonchelate boron guanidinato intermediate.

Carbodiimides as catalysts for the reduction of CO2 with boranes
Chem. Commun., 2018, 54, 4700 DOI: 10.1039/c8cc02139b
Carbodiimides catalyse the reduction of CO2 with H-BBN or BH3– SMe2 to give either mixtures of CH2(OBBN)2 and CH3OBBN or (MeOBO)3 and B(OMe)3 under mild conditions (25–60ºC, 1 atm CO2). Stoichiometric reactions and theoretical calculations were performed to unveil the mechanism of these catalytic processes.
Guanidine Substitutions in Naphthyl Systems to Allow a Controlled Excited-State Intermolecular Proton Transfer: Tuning Photophysical Properties in Aqueous Solution
J. Phys. Chem. C 2018, 122, 9363−9373 DOI: 10.1021/acs.jpcc.8b02576
The excited-state intermolecular proton transfer process (ESPT) in aqueous solution is achieved and controlled by the incorporation of guanidine groups in a fluorescent structure. The bisguanidine under investigation exhibits a dual fluorescence emission with a very high Stokes shifts in water, ≈86 (7890) and 210 (14 500) nm (cm−1), and an excited-stated deprotonation coupled to an intramolecular charge transfer (ICT) process contributes to this emission. The study demonstrates that the emission properties of the different protonation states are strongly dependent on the solvent environment, which also allows luminescence of the molecule to be tuned. The results of this work show the potential utility of guanidine substitution for the stabilization of ESPT−ICT processes in water and allow the subsequent logical design of new stimulus-responsive fluorophores.

Simple ZnEt2 as a catalyst in carbodiimide hydroalkynylation: structural and mechanistic studies
Dalton Trans., 2017, 46, 12923 DOI: 10.1039/C7DT02700A
Expanding the possibilities of the use of simple and available ZnEt2 as a catalyst, the hydroalkynylation of carbodiimides with a variety of alkynes to obtain unsaturated substituted amidines is described in this work. Different stoichiometric studies allow proposing that amidinate complexes are intermediates in this catalytic process, produced by easy activation of the C–H bond of the alkyne and formation of alkynyl derivatives followed by a carbodiimide insertion step. Kinetics studies allowed the generation of a rate law for the hydroalkynylation of N,N′-diisopropylcarbodiimide with phenylacetylene which is second order in [carbodiimide], first order in [catalyst] and zero order in [alkyne], with a negligible PhC![[triple bond, length as m-dash]](http://www.rsc.org/images/entities/char_e002.gif) CH/PhCCD isotopic effect, consistent with a rate-determining state involving carbodiimide insertion. The hydroalkynylation reaction has been coupled with isocyanate (and isothiocyanate) insertion and intramolecular hydroamination to obtain imidazolidin-2-ones (or thione). The structures of different plausible intermediates have been determined by X-ray diffraction studies.
CH/PhCCD isotopic effect, consistent with a rate-determining state involving carbodiimide insertion. The hydroalkynylation reaction has been coupled with isocyanate (and isothiocyanate) insertion and intramolecular hydroamination to obtain imidazolidin-2-ones (or thione). The structures of different plausible intermediates have been determined by X-ray diffraction studies.

Insertion reactions of small unsaturated molecules in the N–B bonds of boron guanidinates
Dalton Trans., 2017,46, 10281-10299 DOI: 10.1039/C7DT02081C
We report here 1,1- and 1,2-insertion reactions of small unsaturated molecules in the N–B bonds of two boron guanidinates, (Me2N)C(NiPr)2BCy2 (1) and {iPr(H)N}C(NiPr){N(p–tBu-C6H4)}BCy2 (2), and two bisboron guanidinates(2–), {iPr(BCy2)N}C(NiPr){N(p–tBu-C6H4)}BCy2 (3) and {iPr(C8H14B)N}C(NiPr){N(p-Me-C6H4)}BC8H14 (4), the latter being prepared for the first time by double deprotonation of the corresponding guanidine with the 9-borabicyclo[3.3.1]nonane dimer, (H-BC8H14)2. Compounds 1–4 easily insert aromatic isonitriles, XylNC (Xyl = 2,6-Me2-C6H3) and (p-MeO-C6H4)NC, to give the expected diazaboroles 5–12, some of them being structurally characterised by X-ray diffraction. Interestingly, the BC8H14 derivatives 11 and 12 are in a fast temperature-dependent equilibrium with the de-insertion products, whose thermodynamic parameters are reported here. A correlation between these equilibria and the puckered heterocyclic structure found in the solid state for 11, and confirmed by DFT calculations, is also established. Reactions of the aforementioned guanidinates with CO are more sluggish or even precluded, and only one product, {iPr(H)N}C{N(p–tBu-C6H4)}(NiPr)(CO)BCy2 (13), could be isolated in moderate yields. The 1,2-insertions of benzaldehyde in compounds 1, 2 and 4 are reversible reactions in all cases, and only one of the insertion products, {iPr(H)N}C{N(p–tBu-C6H4)}(NiPr)(PhHCO)BCy2 (16a), was isolated and diffractrometrically characterised. Likewise, CO2 reversibly inserts into a N–B bond of 2to give {iPr(H)N}C{N(p–tBu-C6H4)}(NiPr)(CO2)BCy2 (19) with a conversion of ca. 9%. In all these equilibria, de-insertion is always favoured upon increasing the temperature.

Synthesis, Characterization, DNA Interactions and Antiproliferative Activity on Glioblastoma of Iminopyridine Platinum (II) Chelate Complexes
Dalton Trans., 2016, 45, 15350 DOI: 10.1039/c6dt02913b
The synthesis of novel dialkylboron guanidinates is reported: the symmetrical compounds, (Me2N)-C(NR)2BR’2 [R = iPr, R’ = Nrb (1); R = Cy, R’ = Nrb (2); R = iPr, R’ = Cy (3); R = R’ = Cy (4); R = 2,6-iPr2-C6H3;R’ = Cy (5); Nrb = exo-2-norbornyl] and the asymmetrically coordinated {iPr(H)N}C(NiPr)(NAr)BCy2 [Ar = Ph (6), 4-Me-C6H4 (7), 4-tBu-C6H4 (8)] were prepared by the salt metathesis method from the appropriate lithium guanidinates and chloroboranes. Moreover, the bis(dicyclohexylboron)guanidinate(−2) {iPr(Cy2B)N}C(NiPr){N(4-tBu-C6H4)}BCy2 (9) was also prepared from the corresponding dilithium guanidinate Li2[{N(4-tBu-C6H4)}C(NiPr)2] and ClBCy2. The structures of compounds 1, 3, 6 and 9 were confirmed by X-ray diffraction and all displayed a chelate coordination of the guanidinate ligand to the BR’2 fragment, the latter displaying an additional BCy2 attached to the exocyclic N atom. Solutions of compounds 1–4 reached an equilibrium with the aminoboranes Me2NBR’2 [R’ = Nrb (10), Cy (11)] and the corresponding carbodiimides, which was slow at 25 °C. The thermodynamic parameters for these equilibria are also reported. The activation parameters for the equilibrium for compound 1 have been calculated after a kinetic study. Compounds 5–8, with one or two N-aryl fragments bound to a B centre, are more robust and need higher temperatures (80 °C) and prolonged times to give similar carbodiimide de-insertion reactions.

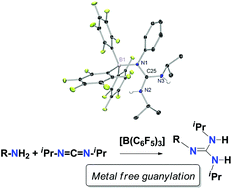
Tris(pentafluorophenyl)borane, [B(C6F5)3], has been used as an efficient catalyst in the guanylation reaction of amines with carbodiimide under mild conditions. A combined approach involving NMR spectroscopy and DFT calculations was employed to gain a better insight into the mechanistic features of this process. The results allowed us to propose a new Lewis acid-assisted Brønsted acidic pathway for the guanylation reaction. The process starts with the interaction of tris(pentafluorphenyl)borane and the amine to form the corresponding adduct, [(C6F5)3B-NRH2] 1, followed by a straightforward proton transfer to one of the nitrogen atoms of the carbodiimide, iPrN=C=NiPr, to produce, in two consequent steps, a guanidine-borane adduct, [(C6F5)3B-NRC(NiPrH)2] 2. The rupture of this adduct liberates the guanidine product RNC(NiPrH)2 3 and interaction with additional amine restarts the catalytic cycle. DFT studies have been carried out in order to study the thermodynamic characteristics of the proposed pathway. Significant borane adducts with amines and guanidines have been isolated and characterized by multinuclear NMR in order to study the N–B interaction and to propose the existence of possible Frustrated Lewis Pairs. Additionally, the molecular structures of significant components of the catalytic cycle, namely 4-tert-butylaniline-[B(C6F5)3] adduct 1b and both free and [B(C6F5)3]-bonded 1-(phenyl)-2,3-diisopropylguanidine, 2a and 3a respectively, have been established by X-ray diffraction.
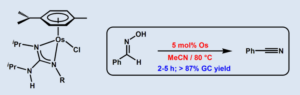
Abstract: The novel guanidinate-osmium(II) complexes [OsCl{k2-(N,N´)-C(NR)(NiPr)-NHiPr}(h6–p-cymene)] [R = Ph (3a), 4-C6H4F (3b), 4-C6H4Cl (3c), 4-C6H4CF3 (3d), 3-C6H4CF3 (3e), 3,5-C6H3(CF3)2 (3f), 4-C6H4CN (3g), 4-C6H4Me (3h), 3-C6H4Me (3i), 2-C6H4Me (3j), 4-C6H4tBu (3k), 2,6-C6H3iPr2 (3l), 2,4,6-C6H2Me3 (3m)] have been synthesized in high yield (70-88%) by treatment of THF solutions of the dimeric precursor [{OsCl(m-Cl)(h6–p-cymene)}2] (1) with four equivalents of the corresponding guanidine (iPrHN)2C=NR (2a-m) at room temperature. The easily separable guanidinium chloride salts [(iPrHN)2C(NHR)][Cl] (4a-m) were also formed in these reactions. The structures of 3a, 3d and 3h were unequivocally confirmed by X-ray diffraction methods. In addition, complexes 3a-m proved to be active in the catalytic dehydration of aldoximes. Best results were obtained with [OsCl{k2-(N,N´)-C(N-4-C6H4CF3)(NiPr)-NHiPr}(h6–p-cymene)] (3d) (5 mol%) which, in acetonitrile at 80 ºC, was able to convert selectively a large variety of aromatic, heteroaromatic, a,b-unsaturated and aliphatic aldoximes into the corresponding nitriles, in high yields and short times.
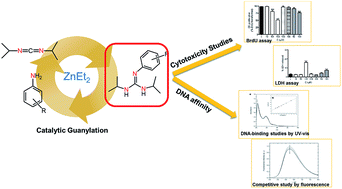
Phenyl-Guanidine Derivatives as Potential Therapeutic Agents for Glioblastoma Multiforme: Catalytic Syntheses, Cytotoxic Effects and DNA Affinity RSC Advances, 2016, 6, 8267. DOI: 10.1039/C5RA17920C
Glioblastoma is a highly malignant form of brain tumor. Current treatment with surgery, temozolamide (TMZ), and radiotherapy only leads to a modest median survival. There is clearly an unmet clinical need for new treatments that are able to arrest the rapid development of the disease through new drugs with antiproliferative activity on glioblastoma cells. In the work described here, several substituted phenyl-guanidine derivatives were developed for application in glioblastoma treatment. The compounds were synthesized by catalytic guanylation reactions and they were fully characterized and assessed for their affinity for DNA by UV titrations and fluorescent intercalator displacement assays. The cytotoxicity levels of the compounds were investigated in the C6 rat glioblastoma cell line by MTT, LDH, and BrdU proliferation assays. Some of the phenyl-guanidine derivatives displayed interesting antitumoral profiles, with a higher potency than the standard drug TMZ in reducing glioblastoma cell proliferation.
Abstract: A combined experimental and quantum chemical study has been performed on rac– and meso-[Zr{1-Me2Si(3-η5-C9H5Et)2}Cl2] (rac– and meso–1) and their hydrogenated forms (rac– and meso–2) to understand ligand effects and guide ligand design for more active ansa-bis(indenyl) zirconocenes for the polymerisation of ethylene. The rac-ansa-zirconocene rac-[Zr(1-Me2Si{3-Et-(η5-C9H9)}2)Cl2] (rac–2) has been prepared and fully characterised by NMR spectroscopy and elemental analysis. The molecular structure of rac–2 has also been determined by single-crystal XRD. The behaviour of the catalysts was analysed in the polymerisation of ethylene and higher activities were obtained for rac–1 and its hydrogenated form rac–2. The influence of the stereochemistry and hydrogenation of the indenyl ligand on the experimental activities has been evaluated by computational studies. The differences along the reaction pathway are dominated by changes in the relative stabilities of the catalytic intermediates. A hybrid density functional B3LYP study, in the presence of toluene as the solvent, indicates that the rac forms give rise to more active species than their meso counterparts. The hydrogenation of the rac forms is a very promising approach to increase activities in polymerisation, in contrast to the meso forms. Finally, the global mechanism rate constants for the polymerisation reaction for each metallocene were calculated by using the thermodynamic formulation of transition-state theory to complement the computational study.
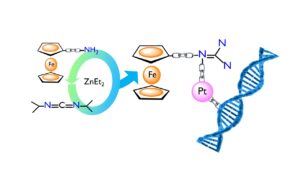
 Catalytically Generated Ferrocene-Containing Guanidines as Efficient Precursors for New Redox-Active Heterometallic Platinum(II) Complexes with Anticancer Activity
Catalytically Generated Ferrocene-Containing Guanidines as Efficient Precursors for New Redox-Active Heterometallic Platinum(II) Complexes with Anticancer Activity
Abstract: The potential of structurally new ferrocene-functionalized guanidines as redox-active precursors for the synthesis of heterometallic platinum(II)-guanidine complexes with anticancer activity was studied. To this end, an atom-economical catalytic approach was followed by using ZnEt2 to catalyze the addition of aminoferrocene and 4-ferrocenylaniline to N,N’-diisopropyl-carbodiimide. Furthermore, reaction of a platinum(II) source with the newly obtained guanidines Fc−N=C(NHiPr)2 (3) and Fc(1,4-C6H4)−N=C(NHiPr)2 (4) provided access to the heterometallic complexes [PtCl2{Fc−N=C(NHiPr)2}(DMSO)] (5), [PtCl2{Fc(1,4-C6H4)−N=C(NHiPr)2}(DMSO)] (6) and [PtCl2{Fc(1,4-C6H4)−N=C(NHiPr)2}2] (7). Electrochemical studies evidence the remarkable electronic effect played by the direct attachment of the guanidine group to the ferrocene moiety in 3, making its one-electron oxidation extremely easy. Guanidine-based Fe–Pt complexes 5 and 6 are active against all human cancer cell lines tested with GI50 values in the range 1.4–2.6 μM, and are more cytotoxic than cisplatin in the resistant T-47D and WiDr cell lines.
 Reactivity of the dimer [{RuCl(µ-Cl)(η3:η3-C10H16)}2] (C10H16 = 2,7-dimethylocta-2,6-diene-1,8-diyl) towards guanidines: Access to novel guanidinate-ruthenium(IV) and ruthenium(II) complexes
Reactivity of the dimer [{RuCl(µ-Cl)(η3:η3-C10H16)}2] (C10H16 = 2,7-dimethylocta-2,6-diene-1,8-diyl) towards guanidines: Access to novel guanidinate-ruthenium(IV) and ruthenium(II) complexes
Abstract: The novel bis(allyl)-ruthenium(IV)-guanidinate complexes [RuCl{κ2-(N,N´)-C(NR)(NiPr)-NHiPr}(η3:η3-C10H16)] (C10H16 = 2,7-dimethylocta-2,6-diene-1,8-diyl; R = Ph (3a), 4-C6H4F (3b), 4-C6H4Cl (3c), 4-C6H4Me (3d), 3-C6H4Me (3e) 4-C6H4tBu (3f)) have been synthesized by treatment of the dimeric precursor [{RuCl(µ-Cl)(η3:η3-C10H16)}2] (1) with 4 equivalents of the corresponding guanidine (iPrHN)2C=NR (2a-f). The easily separable guanidinium chloride salts [(iPrHN)2C(NHR)][Cl] (4a-f) are also formed in these reactions. Attempts to generate analogous Ru(IV)-guanidinate complexes from (iPrHN)2C=NR (R = 2-C6H4Me (2g), 2,4,6-C6H2Me3 (2h), 2,6-C6H3iPr2 (2i)) failed, due probably to the steric hindrance associated with the aryl group in these guanidines. On the other hand, the reaction of dimer [{RuCl(µ-Cl)(η3:η3-C10H16)}2] (1) with (iPrHN)2C=N-4-C6H4C≡N (2j) led to the selective formation of the mononuclear derivative [RuCl2(η3:η3-C10H16){N≡C-4-C6H4-N=C(NHiPr2)2}] (5), in which the guanidine coordinates to ruthenium through the pendant nitrile unit. This result contrasts with that obtained by employing the related Ru(II) dimer [{RuCl(μ-Cl)(η6–p-cymene)}2] (6), whose reaction with 2j afforded the expected guanidinate complex [RuCl{κ2-(N,N´)-C(N-4-C6H4C≡N)(NiPr)-NHiPr}(η6–p-cymene)] (7). Treatment of 7 with dimer 1 yielded a dinuclear Ru(II)/Ru(IV) derivative 8, via cleavage of the chloride bridges of 1 by the C≡N group of 7. Reductive elimination of the 2,7-dimethylocta-2,6-diene-1,8-diyl chain in complexes [RuCl{κ2-(N,N´)-C(NR)(NiPr)-NHiPr}(η3:η3-C10H16)] (3a-f) readily took place in the presence of an excess of 2,6-dimethylphenyl isocyanide, thus allowing the high-yield preparation of the octahedral ruthenium(II) compounds mer-[RuCl{κ2-(N,N´)-C(NR)(NiPr)-NHiPr}(CN-2,6-C6H3Me2)3] (9a-f). The structures of complexes [RuCl{κ2-(N,N´)-C(N-4-C6H4Me)(NiPr)-NHiPr}(η3:η3-C10H16)] (3d), [RuCl{κ2-(N,N´)-C(N-4-C6H4C≡N)(NiPr)-NHiPr}(η6–p-cymene)] (7) and mer-[RuCl{κ2-(N,N´)-C(N-4-C6H4tBu)(NiPr)-NHiPr}(CN-2,6-C6H3Me2)3] (9f), as well as those of the guanidinium chloride salts 4a-c, were unequivocally confirmed by X-ray diffraction methods. In addition, the catalytic behaviour of the guanidinate complexes 3a-f and 9a-f in the redox isomerization of allylic alcohols was also explored.